

Exigences de la norme ISO 15189 laboratoires médicaux version 2022
11/05/2026
Quiz Exigences ISO 15189 version 2022 Vous souhaitez vous familiariser avec la structure de la norme, identifier et comprendre les exigences de l'ISO 15189 version 2022, alors à vous de jouer !
Le quiz "Exigences de l'ISO 15189 version 2022" vous aidera à assimiler les principales exigences de la norme.
Les questions (exigences) de ce quiz sont 164, pas de panique. Les exigences de la norme sont 549 mais ces 164 exigences sont parmi les plus importantes, alors n'hésitez pas à apprendre de façon ludique !
Ne pensez pas que vous pouvez terminer ce quiz en moins d'une heure, voire deux heures, sauf bien sûr si vous êtes un petit génie !
Nouveautés sur la norme laboratoires médicaux ISO 15189 version 2022
Les 459 exigences (doit, doivent, en anglais shall) des articles 4 à 10 et de l'annexe A de l'ISO 15189 version 2022 sont réparties comme il suit :
|
N°
|
Article
|
cycle PDCA
|
Exigences N°
|
Nombre
|
|
4
|
Exigences générales | Planifier (Plan) |
1 à 28
|
28
|
| 5 | Exigences structurelles et de gouvernance | Planifier (Plan) |
29 à 61
|
33 |
| 6 | Ressources | Planifier, Dérouler (Plan, Do) |
62 à 190
|
129
|
| 7 | Processus | Dérouler, Comparer (Do, Check) |
191 à 447
|
257 |
| 8 | Système de management | Planifier, Dérouler, Comparer, Agir (Plan, Do, Check, Act) |
448 à 542
|
95 |
| Annexe A | Exigences EBMD | Planifier, Dérouler, Comparer, Agir (Plan, Do, Check, Act) | 543 à 549 | 7 |
|
Total
|
459
|
|||
.jpg)
Les exigences dans les articles, paragraphes et annexes de la norme ISO 15189 version 2022
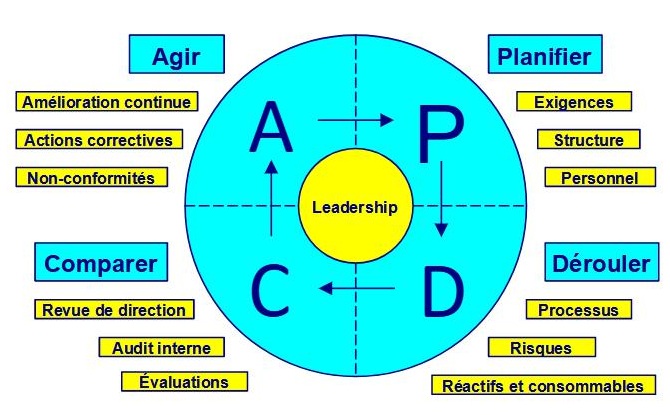
Le cycle PDCA de Deming
Remarque. Toute exigence normalement commence par "Le laboratoire doit ...". Pour simplifier nous présentons les exigences directement en commençant avec le verbe.
|
ISO 15189 - Exigences concernant la qualité et la compétence et commentaires
|
||||
|
N°
|
Paragraphe
|
Exigence
|
Cycle PDCA, liens, commentaires
|
|
|
Exigences générales
|
Planifier (Plan)
|
|||
|
4.1
|
Impartialité
|
|
||
|
1
|
4.1 a
|
Réaliser les activités du laboratoire avec impartialité | Responsabilité de la direction, cf. § 5.2.2 | |
|
2
|
4.1 a
|
Structurer et gérer le laboratoire de manière à préserver l'impartialité | Responsabilité de la direction, cf. § 5.2.2 | |
|
3
|
4.1 b
|
S'engager à exercer ses activités en toute impartialité | Engagement de la direction du laboratoire | |
|
4
|
4.1 c
|
Assumer la responsabilité de ses activités | Par le laboratoire | |
|
5
|
4.1 c
|
Ne pas permettre que des pressions compromettent l'impartialité des activités du laboratoire | Comme des pressions commerciales, financières, sur la propriété, la gouvernance, le management, le personnel ou autres | |
|
6
|
4.1 d
|
Surveiller ses activités et ses relations | Afin d'identifier les menaces qui pèsent sur son impartialité. Responsabilité de la direction, cf. § 5.2.2 | |
|
7
|
4.1 d
|
Inclure les relations avec le personnel | Dans la surveillance des menaces qui pèsent sur son impartialité | |
|
8
|
4.1 e
|
Eliminer ou réduire au minimum l'effet de toute menace | Pouvant peser sur l'impartialité du laboratoire afin de préserver son impartialité | |
|
9
|
4.1 e
|
Pouvoir démontrer la manière dont une menace est limitée | Par le laboratoire | |
|
4.2
|
Confidentialité
|
|
||
|
4.2.1
|
Gestion de l'information
|
|
||
|
10
|
4.2.1
|
Assumer la responsabilité de la gestion de toute information concernant les patients | Informations obtenues au cours de la réalisation des activités du laboratoire | |
| 11 | 4.2.1 | Inclure dans la gestion des informations concernant les patients le respect de la vie privée | Et la confidentialité | |
| 12 | 4.2.1 | Indiquer à l'avance au patient (ou à l'utilisateur) les informations | Qui seront rendues publiques | |
| 13 | 4.2.1 | Traiter toutes les informations comme confidentielles | Sauf celles qui sont rendues publiques (convention entre le laboratoire et le patient) | |
|
4.2.2
|
Communication des informations
|
|
||
|
14
|
4.2.2
|
Informer le patient des informations confidentielles qui seront communiquées | Par le laboratoire qui est tenu de le faire par la réglementation, sauf quand la réglementation l'interdit | |
| 15 | 4.2.2 | Préserver la confidentialité des informations sur le patient qui ne viennent pas de lui | Comme un plaignant, ou autorités réglementaires | |
| 16 | 4.2.2 | Ne pas divulger l'identité de la source des informations | Afin de préserver le respect de la vie privée | |
| 17 | 4.2.2 | Ne pas communiquer l'identité de la source des informations au patient | Sauf si un accord existe avec la source | |
|
4.2.3
|
Responsabilités du personnel
|
|
||
|
18
|
4.2.3
|
Préserver la confidentialité de toutes les informations obtenues par les activités du laboratoire | Cela concerne le personnel, les membres des instances, les contractants, toute personne externe ayant accès au informations du laboratoire | |
|
4.3
|
Exigences relatives aux patients
|
|
||
|
19
|
4.3
|
Veiller à ce que le bien-être, la sécurité et le respect des droits des patients soient prioritaires | Cela concerne la direction du laboratoire | |
| 20 | 4.3 a | Etablir et appliquer le processus Profiter de l'opportunité | De fournir des informations utiles sur le choix des méthodes d'analyse et l'interprétation des résultats d'examens | |
| 21 | 4.3 b | Etablir et appliquer le processus Mettre à disposition des informations de nature publique | Concernant le processus d'examen (coûts et délai de transmission des résultats) | |
| 22 | 4.3 c | Etablir et appliquer le processus Passer en revue périodiquement des examens proposés par le laboratoire | Afin d'assurer que les examens sont appropriés et nécessaires d'un point de vue clinique | |
| 23 | 4.3 d | Etablir et appliquer le processus Communiquer aux parties prenantes des incidents | Préjudiciables pour les patients et des enregistrements des actions mises en place afin de réduire les préjudices | |
| 24 | 4.3 e | Etablir et appliquer le processus Traiter les patients, échantillons et pièces anatomiques |
Avec une attention et un respect rigoureux
|
|
| 25 | 4.3 f | Etablir et appliquer le processus Obtenir un consentement formel |
Quand cela est une exigence
|
|
| 26 | 4.3 g | Etablir et appliquer le processus Tenir disponible l'intégralité des échantillons des patients |
Et les enregistrements en cas de fermeture du laboratoire, rachat ou fusion
|
|
| 27 | 4.3 h | Etablir et appliquer le processus Communiquer les informations au patient ou à un prestataire de soin de santé |
Si demandé par le patient ou d'un établissement agisant en son nom
|
|
| 28 | 4.3 i | Etablir et appliquer le processus Respecter les droits des patients |
Sans aucune discrimination
|
|
|
5
|
Exigences structurelles et de gouvernance
|
Planifier (Plan)
|
||
|
5.1
|
Entité légale
|
|||
| 29 | 5.1 | S'assurer que le laboratoire est une entité légalement responsable |
De ses activités. Un laboratoire gouvernemental est par défaut une entité juridique |
|
|
5.2
|
Directeur de laboratoire
|
|||
|
5.2.1
|
Compétences du directeur de laboratoire
|
|
||
| 30 | 5.2.1 | S'assurer que le laboratoire est dirigé par une personne avec les qualifications spécifiées | Et les compétences et les ressources nécessaires afin de satisfaire aux exigences de l'ISO 15189 version 2022 | |
|
Responsabilités du directeur de laboratoire
|
|
|||
| 31 | 5.2.2 | S'assurer que le directeur de laboratoire est responsable de la mise en place du sysytème de management et de la gestion des risques de toutes les activités du laboratoire | Afin d'identifier et de traiter les risques de prise en charge des patients et de saisir les opportunités d'amélioration | |
| 32 | 5.2.2 | Documenter les missions et responsabilités du directeur de laboratoire | Description de fonction disponible en interne, cf. § 8.4 | |
|
|
5.2.3
|
Délégation des missions et / ou responsabilités
|
|
|
| 33 | 5.2.3 | Documenter les cas de délégation de missions ou de responsabilités par le directeur de laboratoire | A du personnel qualifié et compétent, cf. § 8.4 | |
| 34 | 5.2.3 | S'assurer que la responsabilité ultime est conservée par le directeur de laboratoire | Concernant le fonctionnement général du laboratoire | |
|
5.3
|
Activités du laboratoire
|
|
||
|
5.3.1
|
Généralités
|
|
||
| 35 | 5.3.1 | Spécifier et documenter le champ des activités du laboratoire | Y compris les activités réalisées sur d'autres sites (examens de biologie médicale délocalisée), en conformité avec les exigences de l'ISO 15189 version 2022, cf. § 8.4 | |
| 36 | 5.3.1 | Déclarer la conformité aux exigences de l'ISO 15189 seulement pour les activités du laboratoire | Ce qui exclut les activités externalisées, cf. § 6.8 | |
|
|
5.3.2
|
Conformité aux exigences
|
|
|
| 37 | 5.3.2 | Exécuter toutes les activités du laboratoire de façon à satisfaire aux exigences de l'ISO 15189 version 2022 | Et aussi des utilisateurs, des autorités réglementaires et des organismes d'accréditation | |
|
5.3.3
|
Prestations de conseils
|
|
||
| 38 | 5.3.3 | S'assurer que les prestations de conseils et interprétations appropriées sont disponibles | Et répondent aux besoins des patients et utilisateurs. Responsabilité du directeur de laboratoire | |
| 39 | 5.3.3 a | Établir des dispositions de communication avec les utilisateurs du laboratoire | Concernant les conseils sur le choix et l'utilisation des examens, le type d'échantillons requis, les indications et limitations cliniques des méthodes d'analyse et la fréquence des demandes d'examens | |
| 40 | 5.3.3 b | Établir des dispositions de communication avec les utilisateurs du laboratoire | Afin de fournir des avis professionnels sur l'interprétation des résultats des examens | |
| 41 | 5.3.3 c | Établir des dispositions de communication avec les utilisateurs du laboratoire | Afin de faciliter l'utilisation efficace des examens du laboratoire | |
| 42 | 5.3.3 d | Établir des dispositions de communication avec les utilisateurs du laboratoire | Concernant des conseils dans des domaines scientifiques et ligistiques (comme cas d'échantillons qui ne satisfont pas aux critères d'acceptabilité) | |
|
5.4
|
Structure et autorité
|
|
||
|
5.4.1
|
Généralités
|
|
||
| 43 | 5.4.1 a | Définir son organisation et sa structure | Y compris sa position dans l'organisation mère et ses relations entre la direction et les services techniques et de support (organigramme) | |
| 44 | 5.4.1 b | Spécifier les responsabilités et autorités des personnes qui gèrent, exécutent et vérifient les travaux | Qui ont un impact sur les activités du laboratoire. Ne pas oublier les voies de communication et les relations entre les personnes (descriptions de fonction) | |
| 45 | 5.4.1 c | Spécifier les procédures avec suffisamment de détails | Afin d'assurer une réalisation cohérente des activités du laboratoire et la validité des résultats, cf. § 7.3.3 | |
|
5.4.2
|
Management de la qualité
|
|
||
| 46 | 5.4.2 a | S'assurer que le personnel du laboratoire dispose de l'autorité et des ressources pour réaliser ses missions concernant | La mise en place, le suivi et l'amélioration du système de management (description de fonction) | |
| 47 | 5.4.2 b | S'assurer que le personnel du laboratoire dispose de l'autorité et des ressources pour réaliser ses missions concernant | L'identification des écarts des exigences du système de management et des procédures de réalisation | |
| 48 | 5.4.2 c | S'assurer que le personnel du laboratoire dispose de l'autorité et des ressources pour réaliser ses missions concernant | La mise en place d'actions afin de prévenir des écarts ou les réduire | |
| 49 | 5.4.2 d | S'assurer que le personnel du laboratoire dispose de l'autorité et des ressources pour réaliser ses missions concernant | L'obligation de rendre compte à la direction des performances du système de management et des besoins d'amélioration, cf. §§ 8.6 et 8.9 | |
| 50 | 5.4.2 e | S'assurer que le personnel du laboratoire dispose de l'autorité et des ressources pour réaliser ses missions concernant | L'assurance de l'efficacité des activités du laboratoire | |
|
5.5
|
Objectifs et politiques
|
|
||
| 51 | 5.5 a 1 | Établir et tenir à jour des objectifs et politiques | Afin de répondre aux besoins et attentes des patients et utilisateurs, cf. § 8.2. Responsabilité du directeur de laboratoire | |
| 52 | 5.5 a 2 | Établir et tenir à jour des objectifs et politiques | Afin d'appliquer les bonnes pratiques professionnelles. Responsabilité du directeur de laboratoire | |
| 53 | 5.5 a 3 | Établir et tenir à jour des objectifs et politiques | Afin de réaliser les examens appropriés selon l'utilisation prévue. Responsabilité du directeur de laboratoire | |
| 54 | 5.5 a 4 | Établir et tenir à jour des objectifs et politiques | Afin de se conformer aux exigences de l'ISO 15189 version 2022. Responsabilité du directeur de laboratoire | |
| 55 | 5.5 b | Établir et tenir à jour des objectifs et politiques | Afin de s'assurer que les objectifs sont mesurables et cohérents avec les politiques | |
| 56 | 5.5 b | Établir et tenir à jour des objectifs et politiques | Afin de s'assurer que les objectifs et les politiques sont appliqués à tous les nivaux | |
| 57 | 5.5 c | Établir et tenir à jour des objectifs et politiques | Afin de s'assurer que les modifications planifiées et mises en place préservent l'intégralité du système de management. Responsabilité du directeur de laboratoire | |
| 58 | 5.5 d | Établir et tenir à jour des objectifs et politiques | Afin de définir des indicateurs qualité pour évaluer et surveiller les performances des aspects clés des processus préanalytiques, analytiques et postanalytiques par rapport aux objectifs, cf. § 8.8.2 | |
|
Gestion des risques
|
|
|||
| 59 | 5.6 a | Établir, mettre en place et tenir à jour des processus | Afin d'identifier les risques de préjudices pour les patients et les opportunités d'amélioration à saisir. Responsabilité du directeur de laboratoire. L'ISO 22367 présente plus de détails sur les risques et l'ISO 35001 sur la gestion des biorisques dans les laboratoires médicaux | |
| 60 | 5.6 a | Définir les actions afin de trairer les risques | Et saisir les opportunités d'amélioration, cf. § 8.5. Responsabilité du directeur de laboratoire | |
| 61 | 5.6 b | S'assurer que l'efficacité des processus est évaluée | Et que les processus sont modifiés si ceux-ci sont inéfficaces. Responsabilité du directeur de laboratoire | |
|
6
|
Exigences relatives aux ressources
|
Planifier, Dérouler (Plan, Do)
|
||
|
|
6.1
|
Généralités
|
|
|
| 62 | 6.1 | Disposer du personnel, des installations, des équipements, des réactifs des consommables et des services de support | Nécessaires à la maîtrises des activités du laboratoire | |
|
|
6.2
|
Personnel
|
|
|
|
|
6.2.1
|
Généralités
|
|
|
| 63 | Pouvoir faire appel à un nombre suffisant de personnes compétentes | Afin de réaliser les activités du laboratoire | ||
| 64 | 6.2.1 b | Agir de manière impartiale et éthique, être compétent et satisfaire aux exigences du système de management du laboratoire | Cela concerne le personnel interne et externe du laboratoire qui pourrait avoir une influence sur les activités du laboratoire. L'ISO/TS 22583 présente des recommandations sur les examens de biologie médicale délocalisée | |
| 65 | 6.2.1 c | Communiquer au personnel l'importance du respect des besoins et attentes des utilisateurs | Et des exigences de l'ISO 15189 version 2022 | |
| 66 | 6.2.1 d | Disposer d'un programme d'accueil pour les nouveaux embauchés | Afin de leurs présenter les conditions générales, les locaux, les exigences en matière de santé et de sécurité au travail, livret d'accueil, cf. § 8.4 | |
|
6.2.2
|
Exigences relatives aux compétences
|
|
||
| 67 | 6.2.2 a | Spécifier les compétences pour chaque fonction ayant une influence sur les résultats des activités du laboratoire | Comme niveau d'études, de qualification, de formation, de recyclage professionnel, de connaissances techniques, de savoir-faire et d'expérience, descriptions de fonction, cf. § 8.4 | |
| 68 | 6.2.2 b | S'assurer que le personnel dispose de compétences nécessaires | Afin de réaliser les activités du laboratoire dont il est responsable | |
| 69 | 6.2.2 c | Disposer d'un processus Gérer le personnel |
Qui inclut la fréquence d'évaluation des compétences, cf. § 6.2.2. Exemples de méthodes d'évaluation :
|
|
| 70 | 6.2.2 d | Tenir disponibles des documents sur la compétence du personnel | Cf. §§ 6.2.5 et 8.4 | |
|
6.2.3
|
Autorisation
|
|
||
| 71 | 6.2.3 a | Autoriser des personnes à effectuer des activités particulières | Comme choisir, mettre en place, modifier, valider et vérifier des méthodes | |
| 72 | 6.2.3 b | Autoriser des personnes à effectuer des activités particulières | Comme passer en revue des résultats, éditer et diffuser des comptes rendus | |
| 73 | 6.2.3 c | Autoriser des personnes à effectuer des activités particulières | Comme utiliser des systèmes d'information y compris accès aux données du patient, saisie des données et résultats d'examens | |
|
6.2.4
|
Formation continue et développement professionnel
|
|
||
| 74 | 6.2.4 | Proposer une programme de formation continue | Aux personnes participant aux processus managériaux et technique, cf. § 8.4 | |
| 75 | 6.2.4 | S'assurer que l'ensemble du personnel suit une formation continue | Et à des programmes de développement professionnel | |
| 76 | 6.2.4 | Passer en revue régulièrement l'efficacité des programmes de formation | Et autres activités | |
|
6.2.5
|
Enregistrements relatifs au personnel
|
|
||
| 77 | 6.2.5 a | Tenez à jour des procédures et conservez des enregistrements | Concernant la définition des exigences des compétences du personnel, cf. § 6.2.2 a | |
| 78 | 6.2.5 b | Tenez à jour des procédures et conservez des enregistrements | Concernant les descriptions de fonction, cf. § 6.2.2 a | |
| 79 | 6.2.5 c | Tenez à jour des procédures et conservez des enregistrements | Concernant lla formation et le recyclage professionnel, cf. § 6.2.4 | |
| 80 | 6.2.5 d | Tenez à jour des procédures et conservez des enregistrements | Concernant l'autorisation du personnel, cf. § 6.2.3 | |
| 81 | 6.2.5 e | Tenez à jour des procédures et conservez des enregistrements | Concernant lle suivi des compétences du personnel, cf. § 6.2.2 | |
|
|
6.3
|
Installations et conditions environnementales
|
|
|
|
|
6.3.1
|
Généralités
|
||
| 82 | 6.3.1 | Adapter les installations et conditions environnementales aux activités du laboratoire | Cf. § 5.3. L'ISO 15190 présente des détails sur les installations et conditions environnementales | |
| 83 | 6.3.1 | Ne pas compromettre la validité des résultats ou la sécurité des patients, utilisateurs ou visiteurs |
En adaptant les installations et conditions environnementales aux activités du laboratoire. Exemples de conditions environnementales nuisibles :
|
|
| 84 | 6.3.1 | Appliquer ces exigences aux installations associées aux processus préanalytiques | Et aux autres sites et examens de biologie médicale délocalisée (EBMD), cf. Annexe A | |
| 85 | 6.3.1 | Spécifier, surveiller et enregistrer les exigences relatives aux installations et conditions environnementales | Afin de réaliser les activités du laboratoire, cf. § 8.4 | |
|
|
6.3.2
|
Maîtrise des installations
|
|
|
| 86 | 6.3.2 | Mettre en place, enregistrer, surveiller et passer en revue périodiquement des dispositions | Afin de maîtriser les installations | |
| 87 | 6.3.2 a | Inclure dans ces dispositions | Le contrôle de l'accès aux installations, y compris la sécurité, la confidentialité, la qualité et la protection des informations médicales et des échantillons des patients | |
| 88 | 6.3.2 b | Inclure dans ces dispositions |
La prévention des contaminations, des interférences et autres facteurs néfastes sur les activités du laboratoire comme par des sources :
|
|
| 89 | 6.3.2 c | Inclure dans ces dispositions |
La prévention des contaminations croisées comme quand :
|
|
| 90 | 6.3.2 d | Inclure dans ces dispositions | La mise à disposition d'installations et d'équipements de sécurité et la vérification régulière de leur bon fonctionnement (systèmes d'urgence, alarmes, accéssibilité aux douches d'urgence, équipements de lavage des yeux, matériel de réanimation) | |
| 91 | 6.3.2 e | Inclure dans ces dispositions | Le maintien des installations dans un état fiable | |
|
|
6.3.3
|
Installations de stockage
|
|
|
| 92 | 6.3.3 a | Adapter l'espace et les conditions de stockage | Afin d'assurer l'intégrité permanente des échantillons, des équipements, des réactifs, des consommables et de la documentation | |
| 93 | 6.3.3 b | Entreposer les échantillons des patients et les matériaux utilisés dans les processus analytiques de manière à éviter toute contamination croisée | Et toute détérioration | |
| 94 | 6.3.3 c | S'assurer que les installations de stockage et d'élimination des matériaux dangereux et des déchets biologiques sont appropriées à la classe de dangérosité des matériaux | Et respecter les exigences légales et réglementaires applicables | |
|
|
6.3.4
|
Installations destinées au personnel
|
|
|
| 95 | 6.3.4 | S'assure que le personnel a accès à des toilettes, un point d'eau potable | Et à des installations pour stocker les équipements de protection individuelle et des vêtements | |
| 96 | 6.3.4 | Prévoir un espace pour les activités du personnel | Et une salle d'étude (de réunions) et de repos | |
|
|
6.3.5
|
Installations destinées au prélèvement des échantillons
|
|
|
| 97 | 6.3.5 a | S'assurer que les installations destinées au prélèvement des échantillons | Permettent de prélever les échantillons de manière à ne pas invalider les résultats ou ne pas compromettre la qualité des examens. L'ISO 20658 présente des détails sur les installations pour le prélèvement d'échantillons | |
| 98 | 6.3.5 b | S'assurer que les installations destinées au prélèvement des échantillons | Assurent la confidentialité, le confort et la satisfaction des besoins des patients et l'accueil des accompagnateurs | |
| 99 | 6.3.5 c | S'assurer que les installations destinées au prélèvement des échantillons | Disposent de deux zones distinctes d'accueil des patients et de prélèvement | |
| 100 | 6.3.5 d | S'assurer que les installations destinées au prélèvement des échantillons | Disposent de matériel de premier secours en état de fonctionner pour les patients et le personnel | |
|
|
6.4
|
Equipements
|
|
|
|
|
6.4.1
|
Généralités
|
|
|
| 101 | 6.4.1 | Disposer de processus pour gérer les équipements | Afin d'en assurer le bon fonctionnement et de prévenir toute contamination ou détérioration. La gestion des équipements inclut la sélection, l'acquisition, l'installation, les essais d'acceptation, la manutention, le transport, le stockage, l'utilisation, la maintenance et la mise hors service des équipements. Les équipements sont par exemple les instruments (matériels et logiciels), les systèmes de mesure, d'information, de stockage et de transport | |
|
|
6.4.2
|
Exigences relatives aux équipements
|
|
|
| 102 | 6.4.2 a | Avoir libre accès aux équipements | Afin de réaliser correctement les activités du laboratoire | |
| 103 | 6.4.2 b | S'assurer que les exigences de l'ISO 15189 version 2022 sont satisfaites | Quand les équipements sont utilisés sans le contrôle direct du laboratoire ou sans respecter toutes les spécifications du fabricant | |
| 104 | 6.4.2 c | Identifier chaque élément de l'équipement d'une façon unique | Et conserver un registre des équipements, cf. §§ 6.4.7 et 8.4 | |
| 105 | 6.4.2 d | Entretenir régulièrement les équipements | Et les remplacer afin d'assurer en continu la qualité des résultats | |
|
6.4.3
|
Procédure d'acceptation des équipements
|
|
||
| 106 | 6.4.3 | Vérifier que les équipements sont conformes aux critères d'acceptabilité | Avant leur utilisation | |
| 107 | 6.4.3 | Permettre d'atteindre l'exactitude de mesure ou l'incertitude de mesure afin de fournir un résultat valide | Selon les §§ 7.3.3 et 7.3.4. La vérification des essais d'acceptation des équipements peut reposer sur le certificat d'étalonnage de l'équipement après sa remise en service | |
|
6.4.4
|
Equipements - mode d'emploi
|
|
||
| 108 | 6.4.4 a | Appliquer des mesures de protection appropriées | Afin d'éviter tout déréglage des équipements qui peut invalider les résultats d'examens | |
| 109 | 6.4.4 b | S'assurer que les équipements sont utilisés par des personnes formées | Ayant l'autorité et la compétence requises | |
| 110 | 6.4.4 c | Faire en sorte que les instructions d'utilisation de l'équipement soient disponibles | Pour toute personne ayant le droit d'utiliser l'équipement | |
| 111 | 6.4.4 d | Utiliser l'équipement selon la spécification du fabricant | Sauf si un nouvel usage a été autorisé par le laboratoire | |
|
6.4.5
|
Maintenance et réparation des équipements
|
|
||
| 112 | 6.4.5 a | Établir et appliquer un programme de maintenance préventive des équipements | Selon les instructions du fabricant | |
| 113 | 6.4.5 a | Enregistrer les écarts par rapport aux calendriers | Ou par rapport aux instructions du fabricant, cf. § 8.4 | |
| 114 | 6.4.5 b | Entretenir les équipements en bon état de fonctionnement | Afin de ne présenter aucun risque pour la santé | |
| 115 | 6.4.5 b | Inclure la sécurité électrique, les arrêts d'urgence dans les exigences d'entretien des équipements | Et la manipulation et l'élimination en toute sécurité des matières dangereuses par le personnel autorisé | |
| 116 | 6.4.5 c | Mettre hors service les équipements défectueux | Ou qui ne respectent pas les exigences spécifiées | |
| 117 | 6.4.5 c | Identifier les équipements hors service tant que leur fonctionnement correct n'a pas été démontré | Par une étiquette ou un marquage | |
| 118 | 6.4.5 c | Examiner les impacts du défaut ou de l'écart | Par rapport aux exigences spécifiées | |
| 119 | 6.4.5 c | Mettre en place des actions correctives dans le cas de travaux non conformes | Cf. § 7.5 | |
| 120 | 6.4.5 d | Décontaminer l'équipement, s'il y a lieu avant de l'utiliser, de le réparer, de le metttre hors service dans un endroit approprié | Et fournir les équipements de protection individuelle nécessaires | |
|
6.4.6
|
Signalement des événements indésirables relatifs aux équipements
|
|
||
| 121 | 6.4.6 | Étudier les événements indésirables et accidents causés par des équipements spécifiques | Et les signaler au fabricant, fournisseur ainsi qu'aux autorités réglementaires, si nécessaire | |
| 122 | 6.4.6 | Disposer de procédures afin de répondre à un appel du fabricant ou un autre avis | Et mettre en place les actions requises par le fabricant | |
|
6.4.7
|
Enregistrements relatifs aux équipements
|
|
||
| 123 | 6.4.7 | Conserver les enregistrements pour tout élément d'équipement | Qui peut avoir une influence sur les résultats des activités du laboratoire, cf. § 8.4 | |
| 124 | 6.4.7 a | Inclure dans les enregistrements | Des informations détaillées sur le fabricant, le fournisseur les éléments de l'équipement et le logiciel maison | |
| 125 | 6.4.7 b | Inclure dans les enregistrements | Les dates de réception, essais d'acceptation et de mise en service | |
| 126 | 6.4.7 c | Inclure dans les enregistrements | Les résultats conformes aux critères d'acceptabilité | |
| 127 | 6.4.7 d | Inclure dans les enregistrements | La localisation de l'équipement | |
| 128 | 6.4.7 e | Inclure dans les enregistrements | L'état de l'équipement à sa réception (neuf, utilisé, reconditionné) | |
| 129 | 6.4.7 f | Inclure dans les enregistrements | Les instructions d'utilisation du fabricant | |
| 130 | 6.4.7 g | Inclure dans les enregistrements | Le programme de maintenance préventive | |
| 131 | 6.4.7 h | Inclure dans les enregistrements | Les activités de maintenance réalisées (en interne et par le prestataire de services externe approuvé) | |
| 132 | 6.4.7 i | Inclure dans les enregistrements | Tout dommage, dysfonctionnement, modification ou réparation | |
| 133 | 6.4.7 j | Inclure dans les enregistrements | Résultats datés de performance (rapports, certificats d'étalonnage ou de vérification) | |
| 134 | 6.4.7 k | Inclure dans les enregistrements | L'état de l'équipement (actif, hors service, en quarentaine, obsolète) | |
| 135 | 6.4.7 | Conserver les enregistrements et les rendre disponibles | Pendant toute la durée de vie de l'équipement, cf. § 8.4.3 | |
|
6.5
|
Etalonnage des équipements et traçabilité métrologique
|
|
||
|
6.5.1
|
Généralités
|
|
||
| 136 | 6.5.1 | Spécifier ses exigences d'étalonnage et de traçabilité | Suffisantes afin d'assurer des résultats d'examens cohérents | |
| 137 | 6.5.1 | Inclure des exigences d'étalonnage et de traçabilité métrologique | Concernant les méthodes quantitatives de mesure d'analyte | |
| 138 | 6.5.1 | Spécifier la caractéristique évaluée et les exigences de reproductibilité dans le temps |
Concernant les méthodes qualitatives et semi-qualitatives. Exemples de méthodes ne permettant pas de traçabilité métrologique :
|
|
|
6.5.2
|
Etalonnage des équipements
|
|
||
| 139 | 6.5.2 | Tenir à jour des procédures pour l'étalonnage des équipements | Ayant un impact direct ou indirect sur les résultats des examens | |
| 140 | 6.5.2 a | Inclure dans les procédures | Les conditions d'utilisation et les instructions du fabricant concernant l'étalonnage de l'équipement | |
| 141 | 6.5.2 b | Inclure dans les procédures | Les résultats de la traçabilité métrologique, cf. § 8.4 | |
| 142 | 6.5.2 c | Inclure dans les procédures |
La vérification de :
|
|
| 143 | 6.5.2 d | Inclure dans les procédures | Le résultat de l'état d'étalonnage et la date de réétalonnage | |
| 144 | 6.5.2 e | Inclure dans les procédures | La conservation des mises à jour des dispositions lors de l'utilisation de facteurs de correction pour le réétalonnage | |
| 145 | 6.5.2 f | Inclure dans les procédures | Les modalités afin de gérer les situations pendant lesquelles l'étalonnage était hors limites afin de réduire les risques du fonctionnement du laboratoire et des patients | |
|
6.5.3
|
Traçabilité métrologique des résultats de mesure
|
|
||
| 146 | 6.5.3 a | Établir et conserver la traçabilité métrologique des résultats de mesure | Aa moyen d'une chaîne ininterrompue et documentée d'étalonnages en reliant les résulatst à une référence appropriée. Les informations de la traçabilité peuvent être fournies par un fabricant de systèmes d'analyses | |
| 147 | 6.5.3 b | Assurer que les résultats de mesure sont traçables y compris par le système international d'unités (SI) | Par un étalonnage réalisé par un laboratoire compétent (accrédité ISO 17025) | |
| 148 | 6.5.3 b | Assurer que les résultats de mesure sont traçables y compris par le système international d'unités (SI) | Par des valeurs certifiées de matériaux de référence fournis par un producteur compétent (certifié ISO 17034 ou ISO 15194) | |
| 149 | 6.5.3 c | Appliquer d'autres moyens de prouver la fiabilité des résultats | Comme des résultats de procédures de mesure de référence, de méthodes spécifiés ou de normes faisant consensus par une comparaison appropriée | |
| 150 | 6.5.3 c | Appliquer d'autres moyens de prouver la fiabilité des résultats | Comme mesurage de l'étalon selon une procédure spécifique. L'ISO 17511 présente des informations sur la manière de gérer les compromis de traçabilité métrologique des mesurandes | |
| 151 | 6.5.3 d | Établir la traçabilité à des séquences génétiques de référence | Pour tout examen génétique | |
| 152 | 6.5.3 e | Démontrer la traçabilité pour les méthodes qualitatives | En examinant un matériau connu ou un nombre suffisant d'échantillons préalablement analysés afin de montrer une identification cohérente et si besoin l'intensité de la réaction | |
|
6.6
|
Réactifs et consommables
|
|
||
|
6.6.1
|
Généralités
|
|
||
| 153 | 6.6.1 | Disposer de processus concernant la sélection, l'approvisionnement, la réception, le stockage, les essais d'acceptation des réactifs et consommables | Y compris la gestion des stocks | |
|
6.6.2
|
Réactifs et consommables - Réception et stockage
|
|
||
| 154 | 6.6.2 | Stocker les réactifs et consommablesselon les spécifications du fabricant | Et surveiller les conditions environnementales, si nécessaire | |
| 155 | 6.6.2 | Vérifier que le lieu de réception dispose de capacités adéquates | Afin d'éviter tout dommage ou détérioration, quand le laboratoire ne reçoit pas directement les réactifs et consommables | |
|
6.6.3
|
Réactifs et consommables - Essais d'acceptation
|
|
||
| 156 | 6.6.3 | Vérifier la performance de chaque réactif ou nouvelle formulation de trousse de réactifs ou nouveau lot de fabrication | Avant son utilisation ou la diffusion des résultats. Cf. § 7.3.7.2 pour la comparaison entre nouveau et ancien lot comme preuve d'acceptation | |
| 157 | 6.6.3 | Vérifier la performance des consommables qui peuvent avoir une incidence sur la qualité des examens | Avant de les utiliser. La vérification peut reposer sur le certificat d'analyse du réactif | |
|
6.6.4
|
Réactifs et consommables - Gestion des stocks
|
|
||
| 158 | 6.6.4 | Établir un système de gestion du stock des réactifs | Et des consommables, cf. § 8.4 | |
| 159 |
6.6.4 |
Distinguer les réactifs et consommables acceptés pour utilisation | Et ceux qui n'ont pas été inspectés et acceptés pour utilisation, cf. § 8.4 | |
|
6.6.5
|
Réactifs et consommables - Mode d'emploi
|
|
||
| 160 | 6.6.5 | S'assurer que les modes d'emploi des réactifs sont facilement consultables | Y compris pour les consommables | |
| 161 | 6.6.5 | Utiliser les réactifs et consommables conformément aux spécification du fabricant | S'ils doivent être utilisés pour d'autres applications ils devront passer par une validation des méthodes d'analyse, cf. § 7.7.3 | |
|
6.6.6
|
Réactifs et consommables - Signalement des événements indésirables
|
|
||
| 162 | 6.6.6 | Étudier les événements indésirables et accidents causés par des réactifs ou consommables spécifiques | Et les signaler au fabricant, fournisseur et autorités réglementaire, si besoin | |
| 163 | 6.6.6 | Disposer d'une procédure pour répondre à un appel du fabricant ou un autre avis | Et mettre en place les actions recommandées par le fabricant, cf. § 8.3 | |
|
6.6.7
|
Réactifs et consommables - Enregistrements
|
|
||
| 164 | 6.6.7 | Conserver des enregistrements pour chaque réactif et consommable | En relation avec le niveau de performance des examens, cf. § 8.4 | |
| 165 | 6.6.7 a | Inclure dans ces enregistrements | La dénomination du réactif ou consommable | |
| 166 | 6.6.7 b | Inclure dans ces enregistrements | Le fabricant, les instructions, la référence du lot | |
| 167 | 6.6.7 c | Inclure dans ces enregistrements | La date de réception, l'état à la réception, la date de péremption, la date de première utilisation, et si besoin, la date de mise hors service du réactif ou consommable | |
| 168 | 6.6.7 d | Inclure dans ces enregistrements | L'ptitude initiale et actuelle à l'utilisation du réactif ou consommable | |
| 169 | 6.6.7 | Inclure dans ces enregistrements quand les réactifs sont préparés ou mélangés en interne | Et en plus le personnel ayant participé à la préparation et les dates de préparation et de péremption | |
|
6.7
|
Contrats de prestations
|
|
||
|
6.7.1
|
Contrats avec les utilisateurs du laboratoire
|
|
||
| 170 | 6.7.1 | Disposer d'une procédure pour établir et passer en revue les contrats | Concernant les prestations d'activités du laboratoire, cf. § 8.3 | |
| 171 | 6.7.1 a | S'assurer que la procédure inclut | Les exigences correctement spécifiées | |
| 172 | 6.7.1 b | S'assurer que la procédure inclut | La capacité et les ressources nécessaire pour satisfaire aux exigences | |
| 173 | 6.7.1 c | S'assurer que la procédure inclut | La notification de l'utilisateur des activités spécifiques qui seront transmises à d'autres laboratoires ou consultants | |
| 174 | 6.7.1 | Informer les utilisateurs de toute modification du contrat | Qui peut impacter les résultats d'examens | |
| 175 | 6.7.1 | Conserver des enregistrements des revues incluant d'éventuelles modifications significatives | Cf. § 8.4 | |
|
6.7.2
|
Contrats avec les opérateurs d'EBMD
|
|
||
| 176 | 6.7.2 | S'assurer que les responsabilités et autorités sont spécifiées et communiquées | Concernant les contrats entre le laboratoire et d'aures parties utilisant des examens de biologie médicale délocalisée (EBMD), cf. Annexe A.2 | |
|
6.8
|
Produits et services fournis par des prestataires externes
|
|
||
|
6.8.1
|
Généralités
|
|
||
| 177 | 6.8.1 a | S'assurer que les produits et services, ayant un impact sur les activités du laboratoire, fournis par des prestataires externes, sont appropriés |
Et sont destinés à être intégrés dans les activités du laboratoire. Exemples de services :
|
|
| 178 | 6.8.1 b | S'assurer que les produits et services, ayant un impact sur les activités du laboratoire, fournis par des prestataires externes, sont appropriés | Quand ils sont fournis directement à l'utilisateur | |
| 179 | 6.8.1 c | S'assurer que les produits et services, ayant un impact sur les activités du laboratoire, fournis par des prestataires externes, sont appropriés | Et utilisés pour contribuer au bon fonctionnement du laboratoire | |
| 180 | 6.8.1 | Collaborer avec d'autres départements ou unités | Afin de satisfaire l'exigence concernant les produits et services fournis par des prestataires externes | |
|
6.8.2
|
Laboratoires sous-traitants et consultants
|
|
||
| 181 | 6.8.2 a | Communiquer ses exigences aux laboratoires sous-traitants et consultants qui délivrent des interprétations et des prestations de conseil | Concernant les procédures, analyses, comptes rendus et prestations de conseils | |
| 182 | 6.8.2 b | Communiquer ses exigences aux laboratoires sous-traitants et consultants qui délivrent des interprétations et des prestations de conseil | Concernant la gestion des résultats critiques | |
| 183 | 6.8.2 c | Communiquer ses exigences aux laboratoires sous-traitants et consultants qui délivrent des interprétations et des prestations de conseil | Concernant les qualifications du personnel et les preuves de compétence | |
| 184 | 6.8.2 | S'assurer que les résultats d'analyse transmis par le laboratoire sous-traitant sont communiqués à la personneayant demandé l'analyse | Responsabilité du laboratoire demandeur, sauf indication contraire indiquée dans le contrat | |
| 185 | 6.8.2 | Tenir à jour la liste des laboratoires sous-traitant et des consultants | Cf. § 8.4 | |
|
6.8.3
|
Revue et approbation des produits et services fournis par des prestataires externes
|
|
||
| 186 | 6.8.3 a | Tenir à jour des procédures et conserver des enregistrements concernant | La définition, la revue et l'approbation des exigences du laboratoire pour tous les produits et services fournis par les prestaires exernes, cf. §§ 8.3 et 8.4 | |
| 187 | 6.8.3 b | Tenir à jour des procédures et conserver des enregistrements concernant | La définition des critères de qualification, de sélection, d'évaluation des performances et de réévaluation des prestataires externes | |
| 188 | 6.8.3 c | Tenir à jour des procédures et conserver des enregistrements concernant | La transmission d'échantillons à des laboratoires sous-traitants | |
| 189 | 6.8.3 d | Tenir à jour des procédures et conserver des enregistrements concernant | La garantie que les produits et services fournis par les prestataires externes sont conformes aux exigences du laboratoire ou des exigences de l'ISO 15189 version 2022 avant leur utilisation | |
| 190 | 6.8.3 e | Tenir à jour des procédures et conserver des enregistrements concernant | La mise en place d'actions suite à l'évaluation des performances des prestataires externes | |
|
7
|
Exigences relatives aux processus
|
|||
|
|
7.1
|
Généralités
|
|
|
|
191
|
7.1
|
Identifier les risques relatifs à la prise en charge des patients | Dans les processus préanalytiques, analytiques et postanalytiques | |
|
192
|
7.1
|
Évaluer les risques | Et les réduire autant que possible | |
|
193
|
7.1
|
Communiquer aux utilisateurs le risque résiduel | Si nécessaire | |
|
194
|
7.1
|
Surveiller et évaluer les risques identifiés et l'efficacité des processus pour les gérer | En fonction du potentiel préjudice pour le patient | |
|
195
|
7.1
|
Identifier les opportunités d'amélioration de la prise en charge des patients | Et développer un cadre pour saisir ces opportunités, cf. § 8.5 | |
|
7.2
|
Processus préanalytiques
|
|
||
|
|
7.2.1
|
Généralités
|
|
|
| 196 | 7.2.1 | Disposer de procédures pour les activités préanalytiques | Et tenir à disposition ces procédures pour le personnel concerné, cf. § 8.3. L'ISO 20658 présente des informations sur le prélèvement et le transport des échantillons. Les normes ISO 20186-1 à 3, ISO 20166, L'ISO 20184, L'ISO 23118 et L'ISO 4307 présentent des informations sur les échantillons issus de sources particulières pour les analyses spécifiques | |
|
7.2.2
|
Informations du laboratoire à destination des patients et utilisateurs
|
|
||
| 197 | 7.2.2 | Mettre à disposition des utilisateurs et patients des informations | Appropriées | |
| 198 | 7.2.2 | S'assurer que les informations sont suffisamment détaillées | Afin de fournir aux utilisateurs des données compréhensibles sur le périmètre des activités du laboratoires et ses exigences | |
| 199 | 7.2.2 a | Inclure dans les informations du laboratoire | L'adresse, les heures d'ouverture et les coordonnées de contact | |
| 200 | 7.2.2 b | Inclure dans les informations du laboratoire | Les procédures de demandes d'examens et de prélèvement d'échantillons | |
| 201 | 7.2.2 c | Inclure dans les informations du laboratoire | Le périmètre des activités du laboratoires et le délai de mise à disposition des résultats | |
| 202 | 7.2.2 d | Inclure dans les informations du laboratoire | La disponibilité de prestations de conseil | |
| 203 | 7.2.2 e | Inclure dans les informations du laboratoire | Les exigences sur le consentement des patients | |
| 204 | 7.2.2 f | Inclure dans les informations du laboratoire | Les facteurs connus ayant un impact sur la réalisation de l'examen ou l'interprétation des résultats | |
| 205 | 7.2.2 g | Inclure dans les informations du laboratoire | Les processus de réclamation du laboratoire | |
|
|
7.2.3
|
Demandes d'examens auprès du laboratoire médical
|
|
|
|
|
7.2.3.1
|
Généralités
|
|
|
| 206 | 7.2.3.1 a | Considérer chaque demande d'examen comme un contrat | Quand la demande a été acceptée par le laboratoire | |
| 207 | 7.2.3.1 b | Détailler suffisamment la demande d'examen | Afin d'assurer une traçabilité sans équivoque du patient par rapport à la demande et de l'échantillon | |
| 208 | 7.2.3.1 b | Détailler suffisamment la demande d'examen | L'identité et les cordonnées du demandeur | |
| 209 | 7.2.3.1 b | Détailler suffisamment la demande d'examen | L'identification des examens demandés | |
| 210 | 7.2.3.1 b | Détailler suffisamment la demande d'examen | La mise à disposition de renseignements cliniques, de conseils techniques et d'une interprétation | |
| 211 | 7.2.3.1 c | Fournir les informations sur la demande d'examen | Dans un format ou support appropriés acceptable pour l'utilisateur | |
| 212 | 7.2.3.1 d | Communiquer avec l'utilisateur ou son représentant | Afin de clarifier sa demande | |
|
7.2.3.2
|
Demandes formulées oralement
|
|
||
| 213 | 7.2.3.2 | Disposer d'une procédure de gestion des demandes d'examen formulées oralement | Qui inclut la transmission d'une confirmation écrite de la demande avec un délai donné, cf. § 8.3 | |
|
|
7.2.4
|
Prélèvement et manipulation des échantillons primaires
|
|
|
|
|
7.2.4.1
|
Généralités
|
|
|
| 214 | 7.2.4.1 | Disposer de procédures de prélèvement et de manipulation des échantillons primaires | Cf. § 8.3 | |
| 215 | 7.2.4.1 | Mettre à disposition les informations | Des personnes responsables du prélèvement des échantillons | |
| 216 | 7.2.4.1 | Enregistrer tout écart par rapport aux procédures de prélèvement | Cf. § 8.4 | |
| 217 | 7.2.4.1 | Évaluer, enregistrer et communiquer au personnel concerné | Les risques et l'impact sur la prise en charge du patient en cas d'acceptation ou de refus d'un échantillon, cf. § 8.4 | |
| 218 | 7.2.4.1 | Passer en revue régulièrement les exigences relatives au volume d'échantillon, au dispositif de prélèvement et aux additifs stabilisant | Afin d'assurer la quantité d'échantillon prélèvée suffisante et que les échantillons sont correctement prélevés pour conserver l'intégrité de l'analyte | |
|
7.2.4.2
|
Informations relatives aux activités de pré-prélèvement
|
|
||
| 219 | 7.2.4.2 | Fournir les informations et instructions des activités de pré-prélèvement | Suffisamment détaillées afin d'assurer l'intégralité de l'échantillon, cf. § 8.4 | |
| 220 | 7.2.4.2 a | Inclure dans les informations relatives aux activités de pré-prélèvement | La préparation du patient (instructions au personnel soignant, à ceux qui effectuent le prélèvement et au patient) | |
| 221 | 7.2.4.2 b | Inclure dans les informations relatives aux activités de pré-prélèvement | La nature et la quantité de l'échantillon primaire à prélever, le descriptif du matériel à utiliser, tout additif nécessaire et l'ordre de prélèvement des échantillons | |
| 222 | 7.2.4.2 c | Inclure dans les informations relatives aux activités de pré-prélèvement | Le moment précis auquel le prélèvement sera effectué | |
| 223 | 7.2.4.2 d | Inclure dans les informations relatives aux activités de pré-prélèvement | La communication des informations cliniques pertinentes, la réalisation des analyses ou l'interprétation des résultats (historique de l'administration du médicament) | |
| 224 | 7.2.4.2 e | Inclure dans les informations relatives aux activités de pré-prélèvement | L'étiquetage des échantillons (identification du patient et du site de prélèvement) | |
| 225 | 7.2.4.2 f | Inclure dans les informations relatives aux activités de pré-prélèvement | Les critères d'acceptation et de rejet des échantillons | |
|
|
7.2.4.3
|
Consentement des patients
|
|
|
| 226 | 7.2.4.3 a | Obtenir le consentement du patient | Pour toutes les procédures effectuées sur le patient (le consentement peut être implicite) | |
| 227 | 7.2.4.3 b | Demander une explication plus détaillée et dans certains cas un enregistrement enregistré | Pour des procédures spéciales qui présentent un risque important de complications suite à l'acte, cf. § 8.4 | |
| 228 | 7.2.4.3 c | Effectuer les procédures nécessaires, à condition qu'elles soient dans l'intérêt du patient | Dans le cas d'urgence, quand l'obtention du consentement du patient n'est pas possible | |
|
|
7.2.4.4
|
Instructions relatives aux activités de prélèvement
|
|
|
| 229 | 7.2.4.4 a | Donner des instructions relatives à la vérification de l'identité du patient sur qui l'échantillons primaire a été prélevé | Afin d'assurer la réalisation du prélèvement et le stockage, avant analyse, en toute sécurité, de manière correcte et appropriée sur le plan clinique | |
| 230 | 7.2.4.4 b | Donner des instructions relatives à la vérification (et si pertinent à l'enregistrement) du respect par le patient des exigences préanalytiques (patient à jeun, dernière prise de médicament) | Afin d'assurer la réalisation du prélèvement et le stockage, avant analyse, en toute sécurité, de manière correcte et appropriée sur le plan clinique, cf. § 8.4 | |
| 231 | 7.2.4.4 c | Donner des instructions relatives au prélèvement des échantillons primaires (decriptif du matériel utilisé, additif nécessaire, ordre de prélèvement des échantillons) | Afin d'assurer la réalisation du prélèvement et le stockage, avant analyse, en toute sécurité, de manière correcte et appropriée sur le plan clinique | |
| 232 | 7.2.4.4 d | Donner des instructions relatives à l'étiquetage des échantillons primaires (lien sans équivoque avec le patient) | Afin d'assurer la réalisation du prélèvement et le stockage, avant analyse, en toute sécurité, de manière correcte et appropriée sur le plan clinique | |
| 233 | 7.2.4.4 e | Donner des instructions relatives à l'enregistrement de la personne prélevant l'échantillon primaire, la date et si pertinent l'heure du prélèvement | Afin d'assurer la réalisation du prélèvement et le stockage, avant analyse, en toute sécurité, de manière correcte et appropriée sur le plan clinique | |
| 234 | 7.2.4.4 f | Donner des instructions relatives aux exigences de séparation ou l'aliquotage de l'échantillon primaire | Afin d'assurer la réalisation du prélèvement et le stockage, avant analyse, en toute sécurité, de manière correcte et appropriée sur le plan clinique | |
| 235 | 7.2.4.4 g | Donner des instructions relatives aux conditions de stockageavant transfert des échantillons | Afin d'assurer la réalisation du prélèvement et le stockage, avant analyse, en toute sécurité, de manière correcte et appropriée sur le plan clinique | |
| 236 | 7.2.4.4 h | Donner des instructions relatives à l'élimination sécurisée des matériaux utilisés | Afin d'assurer la réalisation du prélèvement et le stockage, avant analyse, en toute sécurité, de manière correcte et appropriée sur le plan clinique | |
|
7.2.5
|
Transport des échantillons
|
|
||
| 237 | 7.2.5 a 1 | Donner des instructions pour le conditionnement des échantillons pour le transport | Afin d'assurer le transport des échantillons dans des délais appropriés et en toute sécurité | |
| 238 | 7.2.5 a 2 | Donner des instructions pour assurer que le délai entre prélèvement et réception au laboratoire est approprié | Afin d'assurer le transport des échantillons dans des délais appropriés et en toute sécurité | |
| 239 | 7.2.5 a 3 | Donner des instructions pour le maintien de l'intervalle de température requis (prélèvement et manipulation) | Afin d'assurer le transport des échantillons dans des délais appropriés et en toute sécurité | |
| 240 | 7.2.5 a 4 | Donner des instructions pour le respect de toute exigence spécifique pour assurer l'intégralité des échantillons (utilisation d'additifs stabilisants) | Afin d'assurer le transport des échantillons dans des délais appropriés et en toute sécurité | |
| 241 | 7.2.5 b | Informer immédiatement l'organisation responsable du transport | Lorsque l'intégrité d'un échantillon est compromise ou en cas de risque sanitaire | |
| 242 | 7.2.5 b | Prendre des mesures afin de réduire le risque et éviter que cela se reproduise | Lorsque l'intégrité d'un échantillon est compromise ou en cas de risque sanitaire | |
| 243 | 7.2.5 c | Établir et évaluer périodiquement l'adéquation des systèmes de transport | Des échantillons, cf. § 8.4 | |
|
|
7.2.6
|
Réception des échantillons
|
|
|
|
7.2.6.1
|
Procédures de réception des échantillons
|
|
||
| 244 | 7.2.6.1 a | Inclure dans la procédure de réception des échantillons | La traçabilité des échantillons (demande, étiquetage, patient et s'il y a lieu site anatomique) | |
| 245 | 7.2.6.1 b | Inclure dans la procédure de réception des échantillons | Les critères d'acceptation et de rejet des échantillons | |
| 246 | 7.2.6.1 c | Inclure dans la procédure de réception des échantillons | L'enregistrement de la date (et lh'heure si cela est pertinent) de réception de l'échantillon | |
| 247 | 7.2.6.1 d | Inclure dans la procédure de réception des échantillons | La personne qui reçoit l'échantillon (lorsque cela est pertinent) | |
| 248 | 7.2.6.1 e | Inclure dans la procédure de réception des échantillons | L'évaluation des échantillons par du personnel autorisé (afin de s'assurer de leur conformité aux critères d'acceptabilité) | |
| 249 | 7.2.6.1 f | Inclure dans la procédure de réception des échantillons | Les instructions pour les échantillons urgents (étiquetage spécifique, transport, prise en charge de l'urgence, délais d'obtention, critères spécifiques du compte rendu) | |
| 250 | 7.2.6.1 g | Inclure dans la procédure de réception des échantillons | La garantie que toutes les aliquotes de l'échantillon sont traçables jusqu'à l'échantillon d'origine | |
|
|
7.2.6.2
|
Exceptions relatives à l'acceptation des échantillons
|
|
|
| 251 | 7.2.4.2 a 1 | Utiliser des processus permettant d'assurer la meilleure prise en charge du patient pour un échantillon non conforme | Du fait d'une identification incorrecte (du patient ou de l'échantillon) | |
| 252 | 7.2.4.2 a 2 | Utiliser des processus permettant d'assurer la meilleure prise en charge du patient pour un échantillon non conforme | Du fait d'une instabilité de l'échantillon (retard de transport) | |
| 253 | 7.2.4.2 a 3 | Utiliser des processus permettant d'assurer la meilleure prise en charge du patient pour un échantillon non conforme | Du fait d'une température incorrecte (de stockage ou de manipulation) de l'échantillon | |
| 254 | 7.2.4.2 a 4 | Utiliser des processus permettant d'assurer la meilleure prise en charge du patient pour un échantillon non conforme | Du fait de l'utilisation d'un matériel de recueili napproprié de l'échantillon | |
| 255 | 7.2.4.2 a 5 | Utiliser des processus permettant d'assurer la meilleure prise en charge du patient pour un échantillon non conforme | Du fait d'un volume insuffisant de l'échantillon | |
| 256 | 7.2.4.2 b | Indiquer, après étude du risque pour la sécurité du patient, dans le compte rendu la nature de l'anomalie en cas d'acceptation d'un échantillon dont l'altération a un impact clinique important ou d'un échantillon irremplaçable | Et le cas échéant, conseiller une certaine prudence quant à l'interprétation des résultats | |
|
|
7.2.7
|
Manipulation préanalytique, préparation et stockage
|
|
|
|
7.2.7.1
|
Protection des échantillons
|
|
||
| 257 | 7.2.7.1 | Établir des procédures et disposer d'installations appropriées pour sécuriser les échantillons des patients, assurer l'intégrité des échantillons | Et éviter toute perte ou dégradation lors de leur manipulation, préparation et stockage | |
|
7.2.7.2
|
Critères relatifs à la demande d'examens complémentaires
|
|
||
| 258 | 7.2.7.2 | Inclure dans les procédures du laboratoire les délais acceptables | Pour l'ajout d'examens sur le même échantillon | |
|
|
7.2.7.3
|
Stabilité des échantillons
|
|
|
| 259 | 7.2.7.3 | Spécifier et surveiller, si cela est pertient, le délai entre le prélèvement de l'échantillon et la réalisayion de l'analyse | En considérant la stabilité de l'analyte dans l'échantillon primaire | |
|
7.3
|
Processus analytiques
|
|
||
|
|
7.3.1
|
Généralités
|
|
|
|
260
|
7.3.1 a
|
Sélectionner et utiliser des méthodes d'analyte validées | Afin d'assurer l'exploitation clinique des résultats d'examen | |
| 261 | 7.3.1 b | Faire référence pour les spécifications de performance de chaque méthode d'analyte à l'utilisation prévue de l'examen | Et à son impact sur la prise en charge du patient | |
| 262 | 7.3.1 c | Tenir à jour les procédures et la documentation associée et garantir leur disponibilité au personnel | Concernant toutes les activités du laboratoire, cf § 8.3 | |
| 263 | 7.3.1 d | Respecter les procédures établies | Cf. § 8.3 | |
| 264 | 7.3.1 d | Enregistrer les personnes intervenant sur les processus analytiques | Y compris les opérateurs d'EBMD, cf. § 8.4 | |
| 265 | 7.3.1 e | Évaluer régulièrement les méthodes d'analyse par le personnel autorisé | Afin d'assurer que ces méthodes sont cliniquement appropriées concernant les demandes reçues | |
|
7.3.2
|
Vérification des méthodes d'analyse
|
|
||
|
266
|
7.3.2 a
|
Disposer d'une procédure permettant de vérifier que le laboratoire peut mettre en place correctement les méthodes d'analyse avant de les appliquer pour la première fois | En s'assurant que les performances exigées (par le fabricant ou attendues pour la méthode) seront atteints | |
| 267 | 7.3.2 b | S'assurer que les spécifications confirmées de vérification de performance de la méthode d'analyse sont appropriées à l'utilisation prévue | Des résultats d'examens | |
| 268 | 7.3.2 c | S'assurer que l'étendue de la vérification des méthodes d'analyse est suffisante | Afin d'assurer la validité des résultats et une prise de décision clinique | |
| 269 | 7.3.2 d | Passer en revue les résultats de la vérification | Et enregistrer les résultats conformes aux exigences | |
| 270 | 7.3.2 e | Réaliser une nouvelle vérification aussi étendue que nécessaire | Quand une méthode est révisée par l'organisme qui l'a émise | |
| 271 | 7.3.2 f 1 | Conserver les enregistrements relatifs à la vérification effectuée | Incluant les spécifications de performance à atteindre, cf. § 8.4 | |
| 272 | 7.3.2 f 2 | Conserver les enregistrements relatifs à la vérification effectuée | Incluant les résultats obtenus, cf. § 8.4 | |
| 273 | 7.3.2 f 3 | Conserver les enregistrements relatifs à la vérification effectuée | Incluant une déclaration d'atteinte des spécifications de performance et, si ce n'est pas le cas, les actions mises en place, cf. § 8.4 | |
|
7.3.3
|
Validation des méthodes d'analyse
|
|
||
|
274
|
7.3.3 a 1
|
Valider les méthodes d'analyse dans le cas | Méthodes conçues ou mises au point par le laboratoire | |
| 275 | 7.3.3 a 2 | Valider les méthodes d'analyse dans le cas |
Méthodes appliquées en dehors de leur périmètre prévu comme :
|
|
| 276 | 7.3.3 a 3 | Valider les méthodes d'analyse dans le cas | Après modification significative d'une méthode initialement validée | |
| 277 | 7.3.3 b | Valider les méthodes d'analyse | Avec une étendue suffisante | |
| 278 | 7.3.3 b | Confirmer que les exigences spécifiques pour l'utilisation prévue ont été satisfaites | Au moyen de preuves objectives (spécifications de performance) | |
| 279 | 7.3.3 b | S'assurer que l'étendue de la validation d'une méthode d'analyse est suffisante | Afin d'assurer la validité des résultats pour une prise de décision clinique | |
| 280 | 7.3.3 c | Passer en revue les résultats de la validation | Par le personnel autorisé et compétent | |
| 281 | 7.3.3 c | Enregistrer si les résultats sont conformes aux exigences spécifiées | Cf. § 8.4 | |
| 282 | 7.3.3 d | Examiner l'impact clinique | Quand des modifications de la méthode d'analyse validée sont proposées | |
| 283 | 7.3.3 d | Évaluer l'opportunité de mettre en application la méthode modifiée | Quand des modifications de la méthode d'analyse validée sont proposées | |
| 284 | 7.3.3 e 1 | Conserver les enregistrements relatifs à la validation effectuée | Incluant la procédure de validation utilisée, cf. § 8.4 | |
| 285 | 7.3.3 e 2 | Conserver les enregistrements relatifs à la validation effectuée | Incluant les exigences spécifiques relatives à l'utilisation prévue, cf. § 8.4 | |
| 286 | 7.3.3 e 3 | Conserver les enregistrements relatifs à la validation effectuée | Incluant les spécifications de performance de la méthode, cf. § 8.4 | |
| 287 | 7.3.3 e 4 | Conserver les enregistrements relatifs à la validation effectuée | Incluant les résultats obtenus, cf. § 8.4 | |
| 288 | 7.3.3 e 5 | Conserver les enregistrements relatifs à la validation effectuée | Incluant une déclaration de la validité de la méthode, spécifiant l'adéquation à l'utilisation prévue, cf. § 8.4 | |
|
7.3.4
|
Evaluation de l'incertitude de mesure (IM)
|
|
||
|
289
|
7.3.4 a
|
Évaluer et tenir à jour l'incertitude de mesure, si cela est pertinent | Concernant les valeurs quantitatives mesurées | |
| 290 | 7.3.4 a | Comparer et documenter l'incertitude de mesure | Aux spécifications de performance, cf. § 8.4. L'ISO/TS 20914 présente des informations sur l'IM et des exemples | |
| 291 | 7.3.4 b | Passer en revue régulièrement les évaluations | De l'IM | |
| 292 | 7.3.4 c | Documenter la justification de l'absence d'estimation de l'IM | Concernant les procédures d'analyse pour lesquelles une évaluation de l'IM n'est pas possible ou pertinente | |
| 293 | 7.3.4 d | Tenir à disposition les informations concernant l'IM | Pour les utilisateurs du laboratoire qui le demandent | |
| 294 | 7.3.4 e | Prendre en considération les autres sources d'incertitude (variation biologique) | Quand des utilisateurs demandent des précisions sur l'IM | |
| 295 | 7.3.4 f | Estimer l'IM de la grandeur mesuréeen utilisant des échantillons positifs et négatifs représentatifs | Quand le résultat qualitatif d'une analyse repose sur la détermination de données quantitatives et ce résultat est spécifié comme positif ou négatif par rapport à un seuil | |
| 296 | 7.3.4 g | Prendre en compte l'IM des étapes de mesure intermédiaires ou des résultats de CIQ qui produisent des données quantitatives pour les étapes à haut risque du processus | Pour les examens impliquant des résultats qualitatifs, cf. § 7.3.7.2 | |
| 297 | 7.3.4 h | Prendre en compte l'IM lors de la réalisation de la vérification ou de la validation d'une méthode | Lorsque c'est pertinent | |
|
|
7.3.5
|
Intervalles de référence biologiques et limites de décision clinique
|
|
|
|
298
|
7.3.5
|
Définir et communiquer aux utilisateurs les intervalles de référence biologiques et les limites de décision clinique | Lorsque ces intervalles sont nécessaires pour l'interprétation des résultats d'examen | |
| 299 | 7.3.5 a | Définir les intervalles de référence biologiques | Et les limites de décision clinique | |
| 300 | 7.3.5 a | Enregistrer la base sur laquelle les intervalles sont établis en tenant compte du risque pour les patients | Afin de refléter la patientèle du laboratoire. Les valeurs de référence biologique du fabricant peuvent être utilisées si la population de référence des ces valeurs est vérifiée et jugée acceptable par le laboratoire | |
| 301 | 7.3.5 b | Passer en revue régulièrement les intervalles de référence biologiques | Et les limites de décision clinique | |
| 302 | 7.3.5 b | Communiquer aux utilisateurs | Les éventuelles modifications des intervalles de référence biologiques et les limites de décision | |
| 303 | 7.3.5 c | Évaluer et communiquer, si pertinent, les impacts des modifications apportées à une méthode d'analyse ou préanalytique | Sur les intervalles de référence biologiques et les limites de décision clinique associés | |
| 304 | 7.3.5 d | Identifier la caractéristique de l'intervalle de référence biologique | Pour les examens qui identifient la présence ou l'absence d'une caractérisque (analyses génétiques) | |
|
|
7.3.6
|
Documentation des procédures analytiques
|
|
|
|
305
|
7.3.6 a
|
Documenter ses procédures analytiques de manière suffisamment détaillée | Afin d'assurer une application cohérente des activités et la validité des résultats, cf. § 8.3 | |
| 306 | 7.3.6 b | Rédiger les procédures dans une langue comprise par le personnel | Et tenir disponibles les procédures sur place, cf. § 8.3 | |
| 307 | 7.3.6 c | Référer tout document | A la procédure concernée, cf. § 8.3 | |
| 308 | 7.3.6 d | Intégrer les informations issues des modes d'emploi des produits avec des indications suffisamment détaillées | Dand les procédures concernées | |
| 309 | 7.3.6 e | Expliquer aux utilisateurs les conséquences d'une modification validée à une procédure analytique | Modification qui est susceptible d'avoir une incidence sur l'interprétation des résultats | |
| 310 | 7.3.6 f | Maîtriser tous les documents associés au processus analytique | Selon les exigences du paragraphe 8.3 | |
|
|
7.3.7
|
Garantie de la validité des résultats d'examens
|
|
|
|
|
7.3.7.1
|
Généralités
|
|
|
|
311
|
7.3.7.1
|
Disposer d'une procédure de surveillance de la validité des résultats | Cf. paragraphe 8.3 | |
| 312 | 7.3.7.1 | Enregistrer les données de façon à pouvoir détecter les dérives | Et décalages | |
| 313 | 7.3.7.1 | Appliquer des techniques statistiques, si réalisable | Afin d'évaluer les résultats | |
| 314 | 7.3.7.1 | Planifier la surveillance | Et passer en revue la surveillance | |
|
7.3.7.2
|
Contrôle interne de qualité (CIQ)
|
|
||
| 315 | 7.3.7.2 a | Disposer d'une procédure de contrôle interne de qualité (CIQ) | Afin de surveiller en continu la validité des résultats d'examens conformément aux critères spécifiés et de permettre de vérifier l'atteinte de la qualité prévue et d'assurer la validité des résultats pour une prise de décision clinique | |
| 316 | 7.3.7.2 a 1 | Prendre en compte l'exploitation clinique prévue de l'examen | Car les spécifications de performance pour un même mesurande peuvent varier en fonction du contexte clinique | |
| 317 | 7.3.7.2 a 2 | Disposer d'une procédure qui permet de détecter les variations | Selon les lots de réactifs ou détalons de la méthode d'analyse | |
| 318 | 7.3.7.2 a 2 | Disposer d'une procédure qui prévoie qu'un changement de lot du matériau de CIQ n'ait pas lieu le même jour | Ou dans la même série qu'un changement de lot de réactif ou d'étalon | |
| 319 | 7.3.7.2 a 3 | Envisager l'utilisation d'un matériau de CIQ d'une tierce partie (fabricant de l'instrument ou du réactif) | Soit à la place ou en complément | |
| 320 | 7.3.7.2 b | Choisir un matériau de CIQ | Adapté à son utilisation prévue | |
| 321 | 7.3.7.2 b 1 | Inclure dans les facteurs à prendre en compte | Sa stabilité pour l'usage prévu | |
| 322 | 7.3.7.2 b 2 | Inclure dans les facteurs à prendre en compte | La nature de la matrice, aussi proche que possible des échantillons des patients | |
| 323 | 7.3.7.2 b 3 | Inclure dans les facteurs à prendre en compte | Le comportement du matériau de CIQ par rapport à la méthode d'analyse, aussi proche que possible des échantillons des patients | |
| 324 | 7.3.7.2 b 4 | Inclure dans les facteurs à prendre en compte | Le choix du matériau de CIQ pertinent pour l'utilisation clinique, avec des concentrations au niveau ou proches des limites de décision clinique et, si possible, dans l'intervalle de mesure de la méthode d'analyse | |
| 325 | 7.3.7.2 c | Étudier l'utilisation d'autres méthodes de CIQ | Quand le matériau de CIQ approprié est absent | |
| 326 | 7.3.7.2 c 1 | Utiliser d'autres méthodes | Comme une analyse de dérive des résultats des patients (moyenne mobile appliquée aux résultats, un pourcentage d'échantillons inférieur ou supérieur à une valeur donnée ou associée à un diagnostic) | |
| 327 | 7.3.7.2 c 2 | Utiliser d'autres méthodes | Comme une comparaison des résultats avec ceux d'une méthode alternative, validée avec un étalonnage métrologiquement traçable comme spécifié dans l'ISO 17511 | |
| 328 | 7.3.7.2 c 3 | Utiliser d'autres méthodes | Comme une réanalyse d'échantillons conservés de patients | |
| 329 | 7.3.7.2 d | Réaliser le CIQ à une fréquence définie | En relation avec la stabilité et la robustesse de la méthode d'analyse et du risque de préjudice pour le patient dans le cas d'un résultat érroné | |
| 330 | 7.3.7.2 e | Enregistrer les données | Afin de pouvoir détecter des dérives et des décalages, cf. § 8.4 | |
| 331 | 7.3.7.2 e | Appliquer des techniques statistiques, si c'est pertinent | Afin d'évaluer les résultats | |
| 332 | 7.3.7.2 f | Passer en revue les données de CIQ selon des critères d'acceptabilité définis à intervalles réguliers | Dans un délai qui permet de disposer d'une information significative sur les performance en temps réel | |
| 333 | 7.3.7.2 g | Empêcher la communication des résultats des patients | Dans le cas où le CIQ ne répond pas aux critères d'acceptabilité définis | |
| 334 | 7.3.7.2 g 1 | Rejeter les résultats | Dans le cas où les critères d'acceptabilité définis pour le CIQ ne sont pas satisfaits (les résultats contiennent probablement des erreurs cliniquement significatives) | |
| 335 | 7.3.7.2 g 1 | Réanalyser les échantillons provenant des patients | Après correction de l'anomalie, cf. § 7.5 | |
| 336 | 7.3.7.2 g 2 | Évaluer les résultats des échantillons des patients | Provenant des analyses depuis le dernier CIQ satisfaisant | |
|
7.3.7.3
|
Evaluation externe de la qualité (EEQ)
|
|
||
| 337 | 7.3.7.3 a | Surveiller ses performances en ce qui concerne les méthodes d'analyse | Par rapport aux résultats d'autres laboratoires. Comme participation à des programmes d'évaluation externe de la qualité (EEQ), interprétation des résultats d'examens, y compris les méthodes EBMD, cf. Annexe A | |
| 338 | 7.3.7.3 b | Établir une procédure d'inscription, de participation et de réalisation d'EEQ | Pour les méthodes d'analyse utilisées, si pertinent (programmes disponibles), cf. § 8.3 | |
| 339 | 7.3.7.3 c | Traiter les échantillons des évaluations externes de la qualité | Par le personnel qui réalise les procédures préanalytiques, analytiques et postanalytiques | |
| 340 | 7.3.7.3 d 1 | Permettre de vérifier les processus préanalytiques, analytiques et postanalytiques | Concernant le ou les programmes d'EEQ choisis, cf. §§ 7.2, 7.3 et 7.4 | |
| 341 | 7.3.7.3 d 2 | Fournir des échantillons qui imitent les échantillons des patients afin d'obtenir des évaluations cliniquement pertinentes | Concernant le ou les programmes d'EEQ choisis | |
| 342 | 7.3.7.3 d 3 | Respecter les exigences de l'ISO/IEC 17043 | Concernant le ou les programmes d'EEQ choisis | |
| 343 | 7.3.7.3 e | Tenir compte du mode de détermination de la valeur cible retenue | Concernant le ou les programmes d'EEQ choisis | |
| 344 | 7.3.7.3 e 1 | S'assurer que les cibles sont établies | De manière indépendante apr une méthode de réference. Des valeurs consensuelles peuvent être utilisées pour déterminer si les écarts sont dûs au laboratoire ou à la méthode | |
| 345 | 7.3.7.3 e 2 | S'assurer que les cibles sont établies | A partir de données consensuelles | |
| 346 | 7.3.7.3 e 3 | S'assurer que les cibles sont établies | A partir de données consensuelles définies pour le groupe de pairs correspondant à la méthode utilisée | |
| 347 | 7.3.7.3 e 4 | S'assurer que les cibles sont établies | Par un pannel d'experts. Il peut être utile de faire des comparaisons entre les méthodes utilisant des matériaux commutables d'EEQ | |
| 348 | 7.3.7.3 f | Utiliser d'autres méthodes pour surveiller les performances de la méthode d'analyse |
Au cas où le programme d'EEQ est absent ou inadapté. Exemples de méthodes alternatives acceptables :
|
|
| 349 | 7.3.7.3 f | Fournir une justification du choix d'une autre méthode | Et démontrer l'efficacité de cette méthode d'analyse, cf. § 8.4 | |
| 350 | 7.3.7.3 g | Passer en revue les données d'EEQ à intervalles réguliers, par rapport à des critères d'acceptabilités spécifiés et dans un délai donné | Afin de disposer d'une information significative sur les performances en temps réel | |
| 351 | 7.3.7.3 h | Mettre en place une action appropriée incluant l'évaluation de l'impact clinique de la non-conformité venant d'échantillons des patients | Dand les cas où les résultats d'EEQ ne satisfont pas aux critères d'acceptabilité spécifiés, cf. § 8.7 | |
| 352 | 7.3.7.3 i | Passer en revue les résultats d'examens des patients concernés | Quand l'impact clinique est important | |
| 353 | 7.3.7.3 i | Envisager la nécessité d'un amendement | Et informer les utilisateurs, si besoin | |
|
7.3.7.4
|
Comparabilité des résultats d'examens
|
|
||
| 354 | 7.3.7.4 a | Spécifier une procédure permettant d'établir la comparabilité des résultats des échantillons des patients dans les intervalles cliniques appropriés | Dans les cas où des méthodes ou équipements différents sont utilisés pour un même examen ou l'examen est réalisé sur différents sites. Il convient d'utiliser des échantillons des patients pour comparer les méthodes d'analyse | |
| 355 | 7.3.7.4 b | Enregistrer les résultats de comparabilité et les rendre disponibles | Cf. § 8.4 | |
| 356 | 7.3.7.4 c | Passer en revue la comparabilité des résultats | Régulièrement | |
| 357 | 7.3.7.4 d | Évaluer l'impact des différences identifiées | Sur les intervalles de référence biologiques et les dlimites de décision clinique | |
| 358 | 7.3.7.4 d | Prendre des mesures en conséquence | Lorsque des différences sont identifiées | |
| 359 | 7.3.7.4 e | Informer les utilisateurs de toute différence cliniquement significative | Concernant la comparabilité des résultats | |
|
7.4
|
Processus postanalytiques
|
|||
|
7.4.1
|
Compte rendu des résultats
|
|||
|
7.4.1.1
|
Généralités
|
|||
| 360 | 7.4.1.1 a | Transcrire les résultats d'examens dans un compte rendu de manière exacte, claire, non ambiguë | Et conformément aux instructions de la procédure d'examen, cf. § 8.4 | |
| 361 | 7.4.1.1 a | Inclure dans le compte rendu toutes les informations nécessaires | A l'interprétation des résultats | |
| 362 | 7.4.1.1 b | Disposer d'une procédure pour alerter les utilisateurs en cas de retard de communication des comptes rendus d'examens | Sur la base de l'impact du retard sur la prise en charge du patient | |
| 363 | 7.4.1.1 c | Conserver toutes les informations associées aux comptes rendus | Conformément aux exigences du système de management, cf. § 8.4 | |
|
7.4.1.2
|
Revue et diffusion des résultats
|
|||
| 364 | 7.4.1.2 | Passer en revue et approuver les résultats | Avant toute diffusion | |
| 365 | 7.4.1.2 | S'assurer que le personnel autorisé passe en revue les résultats des examens et les évalue par rapport aux résultats du CIQ | Et aux informations cliniques disponibles et aux résultats d'examens précédents | |
| 366 | 7.4.1.2 | Spécifier les responsabilités et procédures relatives aux modalités de diffusion des résultats d'examens | Surtout par quelle personne et à quelle personne, cf. § 8.3 | |
|
7.4.1.3
|
Compte rendu des résultats critiques
|
|||
| 367 | 7.4.1.3 a | Informer l'utilisateur ou tout autre personne autorisée à être informée dès que cela est pertinent selon les informations cliniques disponibles | Quand les résultats d'examens sont dans les limites decisionnelles critiques | |
| 368 | 7.4.1.3 b | Documenter les actions mises en place, la date, l'heure, la personne responsable, la personne alertée, les résultats transmis, la vérification de l'exactitude des informations communiquées | Et les difficultées en relation avec la notification | |
| 369 | 7.4.1.3 c | Disposer d'une procédure de recours pour le personnel | Quand la personne responsable ne peut pas être jointe | |
|
7.4.1.4
|
Considérations spécifiques relatives aux résultats
|
|||
| 370 | 7.4.1.4 a | Rendre toute information des comptes rendus non transmise à l'utilisateur facilement consultable | Cf. §§ 7.4.1.6 et 7.4.1.7 | |
| 371 | 7.4.1.4 b | Communiquer systèmatiquement à l'utilisateur le compte rendu final | Quand les résultats sont transmis sous forme de compte rendu préliminaire | |
| 372 | 7.4.1.4 c | Conserver les enregistrements de tous les résultats communiqués oralement | Y compris les détails de la vérification de l'exactitude des informations communiquées, cf. § 8.4 | |
| 373 | 7.4.1.4 c | S'assurer que ces résultats sont toujours inclus dans un compte rendu | Cf. § 8.4 | |
|
7.4.1.5
|
Sélection, revue, diffusion et compte rendu automatique des résultats
|
|||
| 374 | 7.4.1.5 a | Établir une procédure qui inclut que les critères de sélection, de revue et de diffusion automatiques sont spécifiés, approuvés, consultables et compréhensible pour le personnel autorisant la diffusion des résultats | Quand le laboratoire utilise un système automatique de sélection, revue, diffusion et compte rendu des résultats, cf. § 8.3 | |
| 375 | 7.4.1.5 b | Établir une procédure qui inclut que les critères sont validés et approuvés avant utilisation, revus et vrifiés en cas de modification du système automatique | Quand le laboratoire utilise un système automatique de sélection, revue, diffusion et compte rendu des résultats, cf. § 8.3 | |
| 376 | 7.4.1.5 c | Établir une procédure qui inclut que les résultats sélectionnés par le système automatique pour une revue non automatique sont identifiables | Quand le laboratoire utilise un système automatique de sélection, revue, diffusion et compte rendu des résultats, cf. § 8.3. Si besoin la date, l'heure de la sélection et de la revue et la personne ayant réalisé la revue sont consultables | |
| 377 | 7.4.1.5 d | Établir une procédure qui inclut que, si besoin, la sélection, la revue, la diffusion et l'édition de compte rendu automatiques peuvent être suspendus rapidement | Quand le laboratoire utilise un système automatique de sélection, revue, diffusion et compte rendu des résultats, cf. § 8.3 | |
|
7.4.1.6
|
Exigences relatives aux comptes rendus
|
|||
| 378 | 7.4.1.6 a | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | Sur chaque page l'identification du patient, la date de prélèvement de l'échantillon primaire et la date du compte rendu | |
| 379 | 7.4.1.6 b | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | Le laboratoire emetteur du compte rendu | |
| 380 | 7.4.1.6 c | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | Le nom ou autre identifiant de l'utilisateur | |
| 381 | 7.4.1.6 d | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | Le type d'échantillon primaire et toutes les informations spécifiques à l'échantillon (nature, description macroscopique) | |
| 382 | 7.4.1.6 e | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | L'identification claire des examens réalisés | |
| 383 | 7.4.1.6 f | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | Les méthodes d'analyse utilisées, et si pertinent l'identification harmonisée du mesurande et du principe de mesure | |
| 384 | 7.4.1.6 g | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | Les résultats d'examens en unités de mesure SI, si approprié, ou traçables SI ou autres unités | |
| 385 | 7.4.1.6 h | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | Les intervalles de référence biologiques, les limites de décision clinique, les rapports de probabilité, les diagrammes avec les limites de décision clinique définies | |
| 386 | 7.4.1.6 i | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | L'identification des examens réalisés dans le cadre d'un programme de recherche ou de développement, sans disposition d'indication sur les performances de mesure | |
| 387 | 7.4.1.6 j | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | La personne ayant passé en revue les résultats et ayant autorisé la communication du compte rendu (si absente cette information doit être aisément accessible) | |
| 388 | 7.4.1.6 k | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | L'identification de tous les résultats considérés comme primaires | |
| 389 | 7.4.1.6 l | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | La mise en exergue des résultats critiques | |
| 390 | 7.4.1.6 m | Inclure dans tout compte rendu, sauf si des raisons valables d'omettre des informations sont documentées | Une identification unique de compte rendu complet, indication claire de fin (numéro de page sur total de pages) | |
|
7.4.1.7
|
Informations supplémentaires pour les comptes rendus
|
|||
| 391 | 7.4.1.7 a | Inclure l'heure du prélèvement de l'échantillon primaire | Si cela est nécessaire pour la prise en charge du patient | |
| 392 | 7.4.1.7 b | Tenir consultable la date de communication du compte rendu | Si cette date ne figure pas dans le compte rendu | |
| 393 | 7.4.1.7 c | Tenir disponible l'identification des examens réalisés par le laboratoire sous-traitant | Y compris les informations sans modification des consultants et le laboratoire réalisant les examens | |
| 394 | 7.4.1.7 d 1 | Inclure dans le compte rendu, si pertinent, l'interprétation des résultats et commentaires |
Concernant la qualité et la nature de l'échantillon potentiellement responsables d'une interprétation erronée des résultats d'examens |
|
| 395 | 7.4.1.7 d 2 | Conserver toutes les informations associées aux comptes rendus | Concernat les différences entre les résultats (examens réalisés avec d'autres méthodes ou par d'autres sites | |
| 396 | 7.4.1.7 d 3 | Conserver toutes les informations associées aux comptes rendus | Concernant le risque d'erreur d'interprétation causé par l'utilisation de plusieurs unités de mesure | |
| 397 | 7.4.1.7 d 4 | Conserver toutes les informations associées aux comptes rendus | Concernant l'évolution des résultats ou variations significatives pendant une période donnée | |
|
7.4.1.8
|
Amendements aux comptes rendus des résultats
|
|||
| 398 | 7.4.1.8 a | S'assurer que des procédures de diffusion de résultats révisés ou modifiés incluent l'enregistrement du motif de la modification | Et son inclusion dans le compte rendu révisé | |
| 399 | 7.4.1.8 b | S'assurer que des procédures de diffusion de résultats révisés ou modifiés incluent la fourniture des résultats révisés sous forme d'un document supplémentaire ou d'un transfert de données supplémentaires | Et sont clairement identifiés comme ayant été révisés. Sont inclus aussi la date et l'identité du patient du compte rendu initial | |
| 400 | 7.4.1.8 c | S'assurer que des procédures de diffusion de résultats révisés ou modifiés incluent l'information de la révision | A l'utilisateur | |
| 401 | 7.4.1.8 d | S'assurer que des procédures de diffusion de résultats révisés ou modifiés incluent pour l'édition d'un nouveau compte rendu complet | Une identification unique et la mention du compte rendu initial remplacé | |
| 402 | 7.4.1.8 e | S'assurer que des procédures de diffusion de résultats révisés ou modifiés incluent un enregistrement conservé de ces révisions | Quand les révisions ne peuvent pas être saisies dans le système d'édition de compte rendu | |
|
7.4.2
|
Traitement postanalytique des échantillons
|
|||
| 403 | 7.4.2 | Spécifier la durée et les conditions de conservation des échantillons | Après analyse, cf. § 8.4 | |
| 404 | 7.4.2 a | S'assurer qu'après analyse | L'identité du patient et la nature de l'échantillon sont conservés, § 8.4 | |
| 405 | 7.4.2 b | S'assurer qu'après analyse | La possibilité de soumettre l'échantillon à des examens supplémentaires est connue | |
| 406 | 7.4.2 c | S'assurer qu'après analyse | L'échantillon est conservé de manière à pouvoir réaliser des examens supplémentaires | |
| 407 | 7.4.2 d | S'assurer qu'après analyse | L'échantillon est localisé et peut être récupéré | |
| 408 | 7.4.2 e | S'assurer qu'après analyse | L'échantillon sera éliminé de manière appropriée | |
|
7.5
|
Travaux non conformes
|
|||
| 409 | 7.5 | Disposer d'un processus pour les cas où un aspect des activités du laboratoire ou des résultats d'examens n'est pas conforme | A ses propres procédures, aux spécifications de qualité ou aux exigences des utilisateurs | |
| 410 | 7.5 a | S'assurer que le processus inclut | Les responsabilités et autorités pour la gestion des travaux non conformes | |
| 411 | 7.5 b | S'assurer que le processus inclut | Des actions immédiates et à long terme basées sur l'analyse des risques, cf. §§ 8.5 et 8.7 | |
| 412 | 7.5 c | S'assurer que le processus inclut | Quand un risque de préjudice pour les patients existe, les examens sont arrêtés et les comptes rendus ne sont pas diffusés | |
| 413 | 7.5 d | S'assurer que le processus inclut | Une évaluation de l'importance des travaux non conformes est réalisée y compris l'analyse de l'impact des résultats d'examens diffusés avant l'identification de la non-conformité | |
| 414 | 7.5 e | S'assurer que le processus inclut | La décision d'accepter ou non les travaux non conformes | |
| 415 | 7.5 f | S'assurer que le processus inclut | La révision des résulats d'examens et l'information de l'utilisateur | |
| 416 | 7.5 g | S'assurer que le processus inclut | La responsabilité d'autoriser la reprise des examens | |
| 417 | 7.5 | Mettre en place une action corrective | Proportionnelle à l'impact et la propbabilité de récurrence de la non-conformité, cf. § 8.7 | |
| 418 | 7.5 | Conserver les enregistrements des travaux non conformes et des actions mises en place | Cf. §§ 8.4 et 8.7 | |
|
7.6
|
Maîtrise des données et gestion de l'information
|
|||
|
7.6.1
|
Généralités
|
|||
| 419 | 7.6.1 | Disposer d'un accès aux données et informations nécessaires | Afin de réaliser les activités du laboratoire. L'ISO 22367 présente des détails sur les risques associés aux systèmes informatiques pour les laboratoires de biologie médicale. L'Annexe A de l'ISO 27001 présente des détails sur le référencement des mesures de sécurité de l'information | |
|
7.6.2
|
Autorités et responsabilités concernant la gestion de l'information
|
|||
| 420 | 7.6.2 | S'assurer que les autorités et responsabilités de la gestion des systèmes d'information sont spécifiées | Y compris la maintenance et la modification des systèmes d'information pouvant avoir une incidence sur la prise en charge des patients. La responsabilité en dernier ressort des systèmes d'information du laboratoire revient à la directioncf, §§ 4.2.1 et 5.2.2 | |
|
7.6.3
|
Gestion des systèmes d'information
|
|||
| 421 | 7.6.3 a | Valider le système d'information par le fournisseur et le vérifier par le laboratoire avant sa mise en service y compris sa compatibilité avec d'autres systèmes du laboratoire |
Le système d'information concernant les données et les informations des examens inclut :
|
|
| 422 | 7.6.3 a | Autoriser, documenter et valider tout changement au système |
Y compris les modifications concernant la configuration logicielle ou un logiciel commercial de série |
|
| 423 | 7.6.3 b | Documenter le système d'information |
Et le tenir facilement disponible aux utilisateurs autorisés, y compris le fonctionnement journalier |
|
| 424 | 7.6.3 c | Mettre en place le système d'information |
Y compris en tenant compte de la cybersécurité afin de protéger le système de tout accès non autorisé, d'altération ou perte de données |
|
| 425 | 7.6.3 d | Utiliser le système d'information dans un environnement conforme aux spécifications du fournisseur |
Ou dans le cas de système non informatisé, préserver l'exactitude de l'enregistrement manuel et de sa transcription |
|
| 426 | 7.6.3 e | Maintenir le système d'information afin d'assurer l'intégrité des données et informations |
Et inclure l'enregistrement des défaillances du système et des actions correctives mises en place, cf. § 8.7 |
|
| 427 | 7.6.3 | Contrôler les calculs et transferts de données |
De manière systématique et appropriée |
|
|
7.6.4
|
Plans en cas de panne
|
|||
| 428 | 7.6.4 | Disposer de processus planifiés pour maintenir l'activité en cas de défaillance du système d'information | Pouvant affecter les activités du laboratoire. Y compris la sélection et l'édition de comptes rendus automatiques des résultats. Cf. § 7.8 | |
|
7.6.5
|
Gestion hors site
|
|||
| 429 | 7.6.5 | S'assurer que le prestataire externe satisfait les exigences applicable de l'ISO 15189 version 2022 | Quand le système d'information du laboratoire est l'objet d'une gestion et maintenance hors site | |
|
7.7
|
Réclamations
|
|||
|
7.7.1
|
Processus
|
|
||
| 430 |
7.7.1 | Disposer d'un processus traiter les réclamations | Cf. § 8.6.2 | |
| 431 | 7.7.1 a | Inclure dans le processus traiter les réclamations | Une description des activités de réception, d'évaluation du bien-fondé, d'examen et de prise de décision (actions à entreprendre et opportunités à saisir), cf. §§ 8.7.1 et 8.6 | |
| 432 | 7.7.1 b | Inclure dans le processus traiter les réclamations | Le suivi et l'enregistrement de la réclamation et des actions entreprises, cf. §§ 8.4 et 8.7.1 | |
| 433 | 7.7.1 c | Inclure dans le processus traiter les réclamations | La garantie que l'action a été mise en place et son efficacité, cf. § 8.7.1 | |
| 434 | 7.7.1 | Tenir la description du processus traiter les réclamations | A disposition des utilisateurs | |
|
7.7.2
|
Réception des réclamations
|
|
||
| 435 | 7.7.2 a | Confirmer que la réclamation est liée aux activité de laboratoire | Dès la réception de la réclamation | |
| 436 | 7.7.2 a | Traiter la réclamation | Afin d'y apporter une réponse, cf. § 8.7.1 | |
| 437 | 7.7.2 b | Assumer sa responsabilité | Concernant le recueil des informations afin d'évaluer le bien-fondé de la réclamation | |
| 438 | 7.7.2 c | Accuser réception de la réclamation | Et fournir à la personne ayant porté réclamation les conclusions et, si pertinent, le compte rendu d'avancement de l'état d'avancement | |
|
7.7.3
|
Traitement des réclamations
|
|
||
| 439 | 7.7.3 | Ne pas donner lieu à des actions discriminatoires | Concernant l'examen et le traitement des réclamations | |
| 440 | 7.7.3 | Mener, passer en revue et approuver le traitement des réclamations | Par des personnes non concernées par l'objet de la réclamation | |
| 441 | 7.7.3 | Ne pas compromettre l'impartialité pour toute approche alternative | Quand les ressources sont insuffisantes, cf § 4.1 | |
|
7.8
|
Plan de continuité des activités et de préparation aux situations d'urgence
|
|
||
| 442 | 7.8 | S'assurer que les risques liés aux situations d'urgence sont identifiés | Et qu'un plan de continuité d'activité (PCA) est établi, afin de permettre la poursuite des activités après une interruption, cf. § 7.6.4 | |
| 443 | 7.8 | Tester les plans de continuité d'activité régulièrement | Et la capacité de réponse prévue, lorsque cela est possible | |
| 444 | 7.8 a | Mettre en place une action pour répondre aux situations d'urgence | En prenant en compte les besoins et capacités disponibles du personnel | |
| 445 | 7.8 b | Délivrer les informations nécessaires et une formation | Au personnel concerné, cf. § 6.2.4 | |
| 446 | 7.8 c | Réagir aux situations d'urgences réelles | Cf. § 7.6.4 | |
| 447 | 7.8 d | Entreprendre des actions afin de prévenir ou atténuer les conséquences des situations d'urgence | En tenant compte de l'ampleur de l'urgence et de l'impact potentiel. Plus de détails sont présents dans le CLSI GP36-A (en anglais) | |
|
8
|
Exigences relatives au système de management
|
|||
|
8.1
|
Exigences générales
|
|
||
|
8.1.1
|
Généralités
|
|
||
| 448 | 8.1.1 | Établir, documenter, appliquer et tenir à jour un système de management | Afin d'assurer et démontrer le respect des exigences de l'ISO 15189 version 2022 | |
| 449 | 8.8.1 | Inclure dans le système de management au minimum |
Les points suivants : |
|
|
8.1.2
|
Respect des exigences relatives au système de management
|
|
||
| 450 | 8.1.2 | Assurer et démontrer que le système de management de la qualité respecte les exigences de l'ISO 15189 version 2022 | Inclues dans les articles 4 à 7 et les paragraphes 8.2 à 8.9 | |
|
|
8.1.3
|
Sensibilisation au système de management
|
|
|
| 451 | 8.1.3 a | S'assurer que le personnel effectuant un travail sous le contrôle du laboratoire est sensibilisé | Aux politiques et objectifs du laboratoire, cf. § 5.5 | |
| 452 | 8.1.3 b | S'assurer que le personnel effectuant un travail sous le contrôle du laboratoire est sensibilisé | A sa contribution à l'efficacité du système de management et aux bénéfices de l'amélioration des performances | |
| 453 | 8.1.3 c | S'assurer que le personnel effectuant un travail sous le contrôle du laboratoire est sensibilisé | Aux conséquences du non respect des exigences du système de management | |
|
8.2
|
Documentation du système de management
|
|
||
|
8.2.1
|
Généralités
|
|
||
| 454 | 8.2.1 | Établir, documenter et tenir à jour des politiques et objectifs | Afin de répondre aux objectifs de l'ISO 15189 version 2022. Responsabilité de la direction. Le manuel qualité n'est pas obligatoire et peut être remplacé par une brochure commerciale | |
| 455 | 8.2.1 | S'assurer que les politiques et objectifs sont connus et appliqués | A tous les niveaux du laboratoire, cf. § 5.5 | |
|
8.2.2
|
Compétence et qualité
|
|
||
| 456 | 8.2.2 | Inclure dans les objectifs et politiques la compétence, la qualité | Et la cohérence des activités du laboratoire | |
|
8.2.3
|
Preuves d'engagement
|
|
||
| 457 | 8.2.3 | Fournir la preuve que la direction est engagée sur le développement et l'application du système de management |
Et l'amélioration de son efficacité, cf. § 8.6.1 |
|
|
8.2.4
|
Documentation
|
|
||
| 458 | 8.2.4 | Inclure dans le système de management tous les documents, processus, systèmes et enregistrements relatifs au respect des exigences de l'ISO 15189 version 2022 |
Y être référencés ou avoir un lien au système de management, cf. §§ 8.3 et 8.4. Processus souvent utilisés :
|
|
|
|
8.2.5
|
Accessibilité pour le personnel
|
|
|
| 459 | 8.2.5 | Tenir disponibles au personnel du laboratoire les documents du système de management applicables | Et aussi les informations s'y rapportant | |
|
8.3
|
Maîtrise de la documentation du système de management
|
|
||
|
8.3.1
|
Généralités
|
|
||
| 460 | 8.3.1 | Maîtriser les documents internes et externes |
Liés au respect des exigences de l'ISO 15189 version 2022. Prendre le terme document au sens large. Procédures recommandées :
|
|
|
|
8.3.2
|
Maîtrise des documents
|
|
|
| 461 | 8.3.2 a | S'assurer que les documents sont identifiés | De façon unique (codification) | |
| 462 | 8.3.2 b | S'assurer que l'adéquation des documents a été approuvée avant leur diffusion | Par le personnel autorisé (exepertise et compétences nécessaires) | |
| 463 | 8.3.2 c | S'assurer que les documents sont périodiquement revus | Et mis à jour, si besoin | |
| 464 | 8.3.2 d | S'assurer que les versions pertinentes des documents sont disponibles aux endroits où ils sont utilisés | Et leur diffusion contrôlée, si besoin | |
| 465 | 8.3.2 e | S'assurer que les modifications des documents sont identifiés | Y compris le statut de révision en cours | |
| 466 | 8.3.2 f | S'assurer que les documents sont protégés par rapport aux modifications non autorisées | Y compris tout effacement ou suppression | |
| 467 | 8.3.2 g | S'assurer que les documents sont protégés | Par rapport à un accès non autorisé | |
| 468 | 8.3.2 h | S'assurer que l'utilisation de documents obsolètes est exclue | Y compris que ces documents sont correctement identifiés | |
| 469 | 8.3.2 i | S'assurer qu'une version papier ou électronique du document obsolète est conservée pendant une période définie | Ou conformément aux exigences réglementaires | |
| 8.4 | Maîtrise des enregistrements |
|
||
| 8.4.1 |
Création des enregistrements
|
|
||
| 470 | 8.4.1 | Établir et conserver des enregistrements lisibles |
Afin de démontrer le respect des exigences de l'ISO 15189 version 2022. Enregistrements souvent utilisés :
|
|
| 471 | 8.4.1 | Créer des enregistrements pour chaque activité pouvant avoir une incidence sur la qualité d'un examen | Au moment de la réalisation de l'activité. Un enregistrement peut se présenter sous toute forme et type | |
| 8.4.2 |
Modification des enregistrements
|
|
||
| 472 | 8.4.2 | S'assurer que les modifications des enregistrements peuvent être tracées jusqu'aux versions précédentes | Ou jusqu'aux observations d'origine | |
| 473 | 8.4.2 | Conserver les données et fichiers d'origine et modifiés | Y compris la date, l'heure de la modification, si pertinent, les aspects modifiés et la personne ayant apporté la modification | |
| 8.4.3 |
Conservation des enregistrements
|
|
||
| 474 | 8.4.3 a | Appliquer les procédures nécessaires pour la gestion des enregistrements |
La gestion des enregistrement inclut :
|
|
| 475 | 8.4.3 b | Spécifier la durée de conservation des enregistrements | La durée de conservation peut dépendre des risques identifiés ou de responsabilité légale | |
| 476 | 8.4.3 c | Tenir consultables les comptes rendus des résultats | Aussi longtemps que nécessaire (ou exigé) | |
| 477 | 8.4.3 d | Tenir accessibles tous les enregistrements tout au long de la durée de conservation | Pouvoir être lus quel que soit le support et sont disponibles pour le revue de direction, cf. § 8.9 | |
| 8.5 |
Actions à mettre en oeuvre face aux risques et opportunités d'amélioration
|
|
||
| 8.5.1 |
Identification des risques et opportunités d'amélioration
|
|
||
| 478 | 8.5.1 a | Identifier les risques et opportunités d'amélioration des activités du laboratoire afin de prévenir ou réduire les effets indésirables | Et les défaillances potentielles | |
| 479 | 8.5.1 b | Identifier les risques et opportunités d'amélioration des activités du laboratoire afin de s'améliorer | En saisissant des opportunités d'amélioration | |
| 480 | 8.5.1 c | Identifier les risques et opportunités d'amélioration des activités du laboratoire afin d'assurer que le système de management a atteint les résultats escomptés | Cf. § 5.5 | |
| 481 | 8.5.1 d | Identifier les risques et opportunités d'amélioration des activités du laboratoire afin de réduire les risques concernant la prise en charge des patients | Cf. § 5.6 | |
| 482 | 8.5.1 e | Identifier les risques et opportunités d'amélioration des activités du laboratoire afin de faciliter la mission du laboratoire | Et atteindre ses objectifs, cf. § 5.5 | |
| 8.5.2 |
Actions sur les risques et opportunités d'amélioration
|
|
||
| 483 | 8.5.2 | Hiérarchiser les risques identifiés | Et les maîtriser, cf. § 5.5 | |
| 484 | 8.5.2 | S'assurer que les actions mises en place face aux risques sont proportionnelles à l'impact sur les résultats d'examen | Et la sécurité des patients et du personnel | |
| 485 | 8.5.2 | Enregistrer les décisions prises et les actions mises en place | Face aux risques et aux opportunités d'amélioration | |
| 486 | 8.5.2 | Intégrer et appliquer dans son système de management les actions pour répondre aux risques et opportunités identifiés | Et évaluer leur efficacité. Plus de détails sur les risques sont présents dans l'ISO 22367 et l'ISO 35001 | |
| 8.6 |
Amélioration
|
|
||
| 8.6.1 | Amélioration continue |
|
||
| 487 | 8.6.1 a | Améliorer en continu l'efficacité du système de management conformément aux objectifs et politiques | Y compris les processus préanalytiques, analytiques et postanalytiques, cf. § 5.5 | |
| 488 | 8.6.1 b | Identifier et sélectionner les opportunités d'amélioration | Et développer, documenter et appliquer les actions nécessaires | |
| 489 | 8.6.1 b | Centrer les activités d'amélioration sur les domaines prioritaires suite à l'évaluation des risques | Cf. § 8.5 | |
| 490 | 8.6.1 c | Évaluer l'efficacité des actions entreprises | Cf. § 8.5.2 | |
| 491 | 8.6.1 d | S'assurer que le laboratoire participe aux activités d'amélioration continue | C'est une responsablité de la direction et couvre les domaines pertinents et les résultats de prise en charge des patients | |
| 492 | 8.6.1 e | Communiquer au personnel les plans d'amélioration de la direction | Et les objectifs associés | |
| 8.6.2 |
Retour d'information des patients, des utilisateurs et du personnel
|
|
||
| 493 | 8.6.2 | Solliciter les retours d'information | De la part des patients, des utilisateurs et du personnel | |
| 494 | 8.6.2 | Analyser et exploiter les retours d'information | Afin d'améliorer le système de management, les activités du laboratoire et les prestations pour les utilisateurs | |
| 495 | 8.6.2 | Conserver les enregistrements des retours d'information | Et les actions entreprises | |
| 496 | 8.6.2 | Communiquer au personnel les actions entreprises | Suite au retour d'information du personnel | |
| 8.7 |
Non-conformités et actions correctives
|
|
||
| 8.7.1 |
Actions en cas de non-conformité
|
|
||
| 497 | 8.7.1 a 1 | Répondre à une non-conformité | Et réagir pour la maîtriser | |
| 498 | 8.7.1 a 2 | Répondre à une non-conformité | Et traiter les conséquences, y compris en gérant la sécurité des patients et en informant la personne concernée | |
| 499 | 8.7.1 b | Déterminer les causes de la non-conformité | Trouver la cause première n'est pas toujours évedent | |
| 500 | 8.7.1 c 1 | Évaluer le besoin d'appliquer une action corrective en examinant et analysant la non-conformité | Afin d'éliminer la cause première et de réduire fortement la probabilité de récurrence | |
| 501 | 8.7.1 c 2 | Évaluer le besoin d'appliquer une action corrective en recherchant des non-conformités similaires | Afin d'éliminer la cause première et de réduire fortement la probabilité de récurrence | |
| 502 | 8.7.1 c 3 | Évaluer le besoin d'appliquer une action corrective en évaluant les risques potentiels de la non-conformité | Afin d'éliminer la cause première et de réduire fortement la probabilité de récurrence | |
| 503 | 8.7.1 d | Mettre en place toutes les actions nécessaires | Quand une non-conformité se produit | |
| 504 | 8.7.1 e | Examiner l'action corrective mise en place | Et évaluer son efficacité, cf. § 8.7.2 | |
| 505 | 8.7.1 f | Mettre à jour les risques et opportunités, si besoin | Cf. § 5.6 | |
| 506 | 8.7.1 g | Modifier le système de management | Si cela est pertinent | |
| 8.7.2 |
Efficacité des actions correctives
|
|
||
| 507 | 8.7.2 | S’assurer que les actions correctives sont proportionnelles aux conséquences des non-conformités | Cf. § 8.7.1 a 2 | |
| 508 | 8.7.2 | S’assurer que les actions correctives atténuent les causes identifiées | Cf. § 8.7.1 b | |
| 8.7.3 |
Enregistrement des non-conformités et actions correctives
|
|
||
| 509 | 8.7.3 a | Conserver les enregistrements des non-conformités comme preuves | De la nature des non-conformités, des causes et des actions mises en place, cf. § 8.4 | |
| 510 | 8.7.3 b | Conserver les enregistrements des non-conformités comme preuves | De l'efficacité des actions mises en place | |
| 8.8 |
Evaluations
|
|
||
| 8.8.1 |
Généralités
|
|
||
| 511 | 8.8.1 | Réaliser des évaluations à intervalles planifiés | Afin de démontrer que tous les processus des activités du laboratoires répondent aux exigences des patients, des utilisateurs et de l'ISO 15189 version 2022 | |
| 8.8.2 |
Indicateurs qualité
|
|
||
| 512 | 8.8.2 | Planifier le processus de surveillance des indicateurs qualité |
En incluant l'établissement :
|
|
| 513 | 8.8.2 | Passer en revue les indicateurs régulièrement | Afin de s'assurer de leur adéquation | |
| 8.8.3 |
Audits internes
|
|
||
| 514 | 8.8.3.1 a | Réaliser des audits internes à intervalles planifiés afin de déterminer si le système de management | Est conforme aux exigences du laboratoire (son système de management et ses activités) | |
| 515 | 8.8.3.1 b | Réaliser des audits internes à intervalles planifiés afin de déterminer si le système de management | Est conforme aux exigences de l'ISO 15189 version 2022 | |
| 516 | 8.8.3.1 c | Réaliser des audits internes à intervalles planifiés afin de déterminer si le système de management | Est appliqué et maintenu efficacement | |
| 517 | 8.8.3.2 a | Planifier, établir, appliquer et maintenir un programme d'audit qui inclut | Une priorité aux risques pour les patients causés par les activités du laboratoire | |
| 518 | 8.8.3.2 b | Planifier, établir, appliquer et maintenir un programme d'audit qui inclut |
La prise en compte :
|
|
| 519 | 8.8.3.2 c | Planifier, établir, appliquer et maintenir un programme d'audit qui inclut | Les objectifs, les critères et le périmètre de chaque audit | |
| 520 | 8.8.3.2 d | Planifier, établir, appliquer et maintenir un programme d'audit qui inclut | La sélection des auditeurs (formés et qualifiés) qui sont indépendants de l'activité auditée | |
| 521 | 8.8.3.2 e | Planifier, établir, appliquer et maintenir un programme d'audit qui inclut | La garantie d'objectivité et d'impartialité des activités de l'audit | |
| 522 | 8.8.3.2 f | Planifier, établir, appliquer et maintenir un programme d'audit qui inclut | La communication des résultats au personnes concernées | |
| 523 | 8.8.3.2 g | Planifier, établir, appliquer et maintenir un programme d'audit qui inclut | La mise en place des actions correctives (responsable, délai) | |
| 524 | 8.8.3.2 h | Planifier, établir, appliquer et maintenir un programme d'audit qui inclut | La conservation des enregistrements (respect du programme d'audit, rapport d'audit), cf. § 8.4 | |
| 8.9 |
Revues de direction
|
|
||
| 8.9.1 |
Généralités
|
|
||
| 525 | 8.9.1 | Passer en revue, à intervalles planifiés, le système de management | Afin de s'assurer qu'il est toujours pertinent, adéquat et efficace et respecte les exigences de l'ISO 15189 version 2022 | |
| 8.9.2 |
Eléments d'entrée de la revue
|
|
||
| 526 | 8.9.2 a | Enregistrer les éléments d'entrée de la revue de direction et inclure | Le statut des actions de la revue de direction précédente, les modifications du système de management, les changements des activités et l'adéquation des ressources | |
| 527 | 8.9.2 b | Enregistrer les éléments d'entrée de la revue de direction et inclure | Le respect des objectifs, des politiques et procédures | |
| 528 | 8.9.2 c | Enregistrer les éléments d'entrée de la revue de direction et inclure | Les résultats des indicateurs des processus, des audits internes, de l'analyse des non-conformités, des actions correctives et des audits externes | |
| 529 | 8.9.2 d | Enregistrer les éléments d'entrée de la revue de direction et inclure | Les retours d'information (patients, utilisateurs, personnel) | |
| 530 | 8.9.2 e | Enregistrer les éléments d'entrée de la revue de direction et inclure | L'assurance qualité de la validité des résultats | |
| 531 | 8.9.2 f | Enregistrer les éléments d'entrée de la revue de direction et inclure | L'efficacité des actions face aux risques | |
| 532 | 8.9.2 g | Enregistrer les éléments d'entrée de la revue de direction et inclure | Les performances des prestataires externes, cf. § 6.8 | |
| 533 | 8.9.2 h | Enregistrer les éléments d'entrée de la revue de direction et inclure | Les résultats des comparaisons interlaboratoires | |
| 534 | 8.9.2 i | Enregistrer les éléments d'entrée de la revue de direction et inclure | L'évaluation des activités EBMD, cf. Annexe A | |
| 535 | 8.9.2 j | Enregistrer les éléments d'entrée de la revue de direction et inclure | Les activités de surveillance et de formation, cf. § 6.2.4 | |
| 8.9.3 |
Eléments de sortie de la revue
|
|
||
| 536 | 8.9.3 a | Enregistrer les éléments de sortie de la revue de direction et inclure des décisions et actions | Concernant l'efficacité du système de management et des processus | |
| 537 | 8.9.3 b | Enregistrer les éléments de sortie de la revue de direction et inclure des décisions et actions | Concernant l'amélioration des activités du laboratoire selon les exigences de l'ISO 15189 version 2022 | |
| 538 | 8.9.3 c | Enregistrer les éléments de sortie de la revue de direction et inclure des décisions et actions | Concernant les ressources nécessaires | |
| 539 | 8.9.3 d | Enregistrer les éléments de sortie de la revue de direction et inclure des décisions et actions | Concernant l'amélioration des prestations aux patients et utilisateurs | |
| 540 | 8.9.3 e | Enregistrer les éléments de sortie de la revue de direction et inclure des décisions et actions | Concernant un nouveau changement | |
| 541 | 8.9.3 | S'assurer que les actions décidées en revue de direction sont réalisées | Dans le délai planifié | |
| 542 | 8.9.3 | Communiquer les décisions de la revue de direction au personnel | Cf. § 8.4 | |
| Annexe A |
Exigences supplémentaires relatives aux examens de biologie médicale délocalisée (EBMD)
|
|||
| A.2 |
Gouvernance des EBMD
|
|
||
| 543 | A.10.2 | Assumer la responsabilité de la mise en place des processus pour surveiller l'exactitude et la qualité des EBMD réalisés en interne | De la part de l'organe directeur de l'organisation | |
| 544 | A.10.2 | Spécifier et communiquer les responsabilités et autorités en interne | Concernant les contrats avec les sites réalisant des EBMD | |
| 545 | A.10.2 | Approuver les contrats EBMD sur le plan financier | Et, si pertinent, le plan financier | |
| 546 | A.10.2 | S'assurer que les contrats EBMD concernent le lieu de réalisation des EBMD | Et peuvent être gérés, par exemple, par un comité médical consultatif | |
| A.3 |
Programme d'assurance qualité
|
|
||
| 547 | A.10.3 | Nommer une personne ayant la formation et l'expérience appropriées comme responsable qualité des EBMD | Afin de passer en revue et respecter les exigences de l'ISO 15189 version 2022 concernant les EBMD | |
| A.4 |
Programme de formation
|
|
||
| 548 | A.10.4 | Nommer une personne ayant la formation et l'expérience appropriées pour gérer la formation et l'évaluation des compétences | Du personnel réalisant des EBMD | |
| 549 | A.10.4 | Établir, appliquer et tenir à jour un programme théorique et pratique | Pour le personnel réalisant des EBMD | |
|
|
|
|
|
|

