

Exigences de la norme ISO 13485 version 2016 système de management de la qualité dispositifs medicaux
01/04/2016
Quiz exigences ISO 13485 version 2016 Vous souhaitez vous familiariser avec la structure de la norme, identifier et comprendre les exigences de l'ISO 13485 version 2016, alors à vous de jouer !
Le quiz "Exigences de l'ISO 13485 version 2016" vous aidera à assimiler les principales exigences de la norme.
Les questions (exigences) de ce quiz sont 98, pas de panique. Les exigences de la norme sont 416 mais ces 98 exigences sont parmi les plus importantes, alors n'hésitez pas à apprendre de façon ludique !
Ne pensez pas que vous pouvez terminer ce quiz en moins d'une heure, voire deux heures, sauf bien sûr si vous êtes un petit génie !
Nouveautés de l'ISO 13485 version 2016
Les 416 exigences (doit, doivent, en anglais shall) des articles 4 à 8 de l’ISO 13485 sont réparties comme suit :
|
N°
|
Article
|
cycle PDCA
|
Exigences N°
|
Nombre
|
|
4
|
Système de management de la qualité | Planifier |
1 ÷ 62
|
62
|
|
5
|
Responsabilité de la direction | Planifier, Agir |
63 ÷ 110
|
48
|
|
6
|
Management des ressources | Planifier |
111 ÷ 130
|
20
|
|
7
|
Réalisation du produit | Dérouler |
131 ÷ 325
|
195
|
|
8
|
Mesurage, analyse et amélioration | Constater, Agir |
326 ÷ 416
|
91
|
|
Total
|
416
|
|||
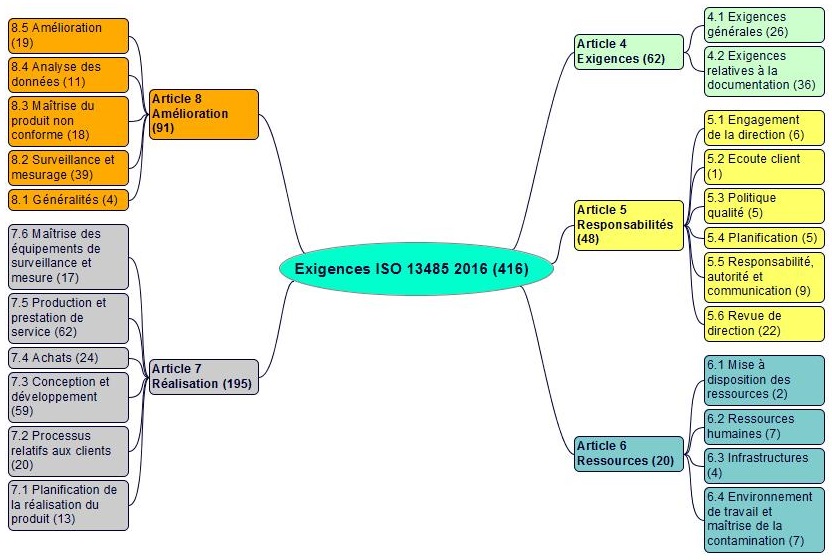
Les exigences et les articles et paragraphes de la norme ISO 13485 version 2016
.gif)
Le cycle PDCA de Deming
Remarques :
- Pour plus de clarté et rester proche de la réalité le terme organisme est remplacé par entreprise
- Pour faciliter la compréhension des exigences le verbe doit (doivent) est remplacé par le verbe le plus proche de l'exigence
- Ressources humaines est remplacé par personnel (comme dans l'ISO 9001 version 2015 édition en anglais)
|
ISO 13485 version 2016 - Exigences et commentaires
|
||||
|
N°
|
Article, paragraphe
|
Exigence
|
Cycle PDCA, liens, commentaires
|
|
|
Système de management de la qualité
|
|
|||
|
|
Exigences générales
|
|
||
|
1
|
4.1.1
|
Documenter un système de management de la qualité (SMQ) | Cf. § 4.2.5 | |
|
2
|
4.1.1
|
Maintenir l'efficacité du SMQ | Efficacité c'est la capacité de réalisation des activités planifiées avec le minimum d'efforts | |
|
3
|
4.1.1 | Établir toute exigence, procédure, activité ou disposition | Exigés par la norme ISO 13485 ou par une exigence réglementaire applicable | |
| 4 | 4.1.1 | Mettre en œuvre toute exigence, procédure, activité ou disposition | Exigés par la norme ISO 13485 ou par une exigence réglementaire applicable | |
| 5 | 4.1.1 | Tenir à jour toute exigence, procédure, activité ou disposition | Exigés par la norme ISO 13485 ou par une exigence réglementaire applicable | |
| 6 | 4.1.1 | Documenter les rôles exercés par l'entreprise dans le cadre des exigences réglementaires applicables | Cf. § 4.2.5. Identifier les rôles de l'entreprise (du fabricant, du fournisseur, du distributeur, du représentant du fabricant) | |
| 7 | 4.1.2 a | Déterminer les processus nécessaires pour le SMQ et l'application de ces processus | Bien que la cartographie des processus ne soit pas explicitement exigée, c’est en pratique la meilleure réponse à cette exigence | |
|
8
|
4.1.2 b
|
Appliquer une approche par les risques pour maîtriser les processus nécessaires au SMQ | Cf. § 7.1. Identifier les risques des processus, leurs niveaux, leurs impacts sur la sécurité et les performances du produit. Justifier le degré choisi de maîtrise du risque | |
|
9
|
4.1.2 c
|
Déterminer la séquence et les interactions de ces processus | Bien que la cartographie des processus ne soit pas explicitement exigée, c’est en pratique la meilleure réponse à cette exigence | |
| 10 | 4.1.3 a | Déterminer les critères et méthodes pour assurer l'efficacité de leur fonctionnement et de leur maîtrise | Pour chaque processus du SMQ | |
| 11 | 4.1.3 b | Assurer la disponibilité des ressources et des informations nécessaires pour soutenir leur fonctionnement et leur surveillance | Pour chaque processus du SMQ | |
| 12 | 4.1.3 c | Mettre en œuvre les actions nécessaires pour obtenir les résultats planifiés et maintenir leur efficacité | Pour chaque processus du SMQ | |
| 13 | 4.1.3 d | Surveiller, mesurer et analyser ces processus | Mesurer selon le cas | |
| 14 |
4.1.3 e
|
Établir et conserver les enregistrements nécessaires pour démontrer la conformité à la norme ISO 13485 et aux exigences réglementaires applicables | Cf. § 4.2.5. Pour chaque processus du SMQ | |
| 15 | 4.1.4 | Gérer les processus du SMQ | Conformément aux exigences de la norme ISO 13485 et aux exigences réglementaires applicables | |
|
16
|
4.1.4 a
|
Évaluer l'incidence sur le SMQ des modifications de ces processus | Décrire la gestion des modifications (règles, critères, suivi) | |
|
17
|
4.1.4 b
|
Évaluer l'incidence sur les dispositifs médicaux dans le cadre de ce SMQ des modifications de ces processus | Cf. § 7.3.9 | |
|
18
|
4.1.4 c
|
Maîtriser les modifications de ces processus | Selon les exigences de la norme ISO 13485 et des exigences réglementaires applicables | |
|
19
|
4.1.5
|
Surveiller et assurer une maîtrise des processus externalisés | Qui peuvent avoir une incidence sur le respect des exigences. Liste des processus externalisés, niveaux de risque associé | |
|
20
|
4.1.5
|
Endosser la responsabilité de conformité pour les processus externalisés | A la norme ISO 13485, aux clients et aux exigences réglementaires applicables | |
|
21
|
4.1.5
|
Garder la maîtrise proportionnée au risque associé | Et à l'aptitude de la partie externe à satisfaire aux exigences d'achats, cf. § 7.4 | |
|
22
|
4.1.5
|
Inclure dans la maîtrise des dispositions écrites relatives à la qualité | Accords qualité fournisseur, surtout pour des processus externalisés avec niveau de risque élevé | |
|
23
|
4.1.6
|
Documenter des procédures pour la validation des applications logicielles utilisées dans le SMQ | Cf. § 4.2.4 | |
|
24
|
4.1.6
|
Valider les applications logicielles avant leur utilisation et après modification | Liste des logiciels validés utilisés dans le SMQ | |
|
25
|
4.1.6
|
Garder l'approche et les activités de validation et revalidation proportionnées au risque associé à l'utilisation des logiciels | Pour chaque logiciel utilisé justifier l'approche de validation par rapport au niveau de risque | |
|
26
|
4.1.6
|
Conserver les enregistrements de ces activités | Cf. § 4.2.5 | |
|
Exigences relatives à la documentation
|
|
|||
|
|
Généralités
|
|
||
|
27
|
4.2.1 a
|
Documenter la politique qualité et les objectifs qualité | Cf. §§ 4.2.4, 4.2.5 . La politique qualité et les objectifs sont formalisés dans un document pertinent et simple | |
| 28 | 4.2.1 b | Inclure sous forme documentée le manuel qualité | Si les procédures ne font pas partie du manuel qualité alors on y fait référence et l’on indique l’endroit où on peut les trouver (cela peut être le système Intranet) | |
|
29
|
4.2.1 c
|
Inclure les procédures documentées et les enregistrements exigés par la norme ISO 13485 | Cf. § 4.2.4. Les exigences de la norme ISO 13485 v 2016 sont 416 | |
| 30 |
4.2.1 d
|
Inclure les documents, y compris les enregistrements, déterminés par l'entreprise comme nécessaires pour assurer la planification, le fonctionnement et la maîtrise efficaces des processus | Cf. § 4.2.5 | |
| 31 | 4.2.1 e | Inclure toute documentation spécifiée par les exigences réglementaires applicables | Cf. § 4.2.4 et 4.2.5. Le strict nécessaire est souvent le meilleur choix | |
|
|
Manuel qualité
|
|||
|
32
|
4.2.2 a
|
Documenter un manuel qualité incluant le domaine d'application du SMQ | Cf. § 4.2.4 | |
| 33 | 4.2.2 b | Documenter un manuel qualité incluant les procédures documentées | Cf. § 4.2.4. Ou incluant une référence aux procédures | |
| 34 | 4.2.2 c | Documenter un manuel qualité incluant une description des interactions des processus du SMQ | Cf. §§ 4.2.1 et 4.2.4 | |
|
35
|
4.2.2
|
Donner un aperçu de la structure de la documentation du SMQ | Le plus simple est la pyramide documentaire : manuel qualité, processus, procédures, documents, enregistrements | |
|
|
Dossier du dispositif médical
|
|
||
|
36
|
4.2.3
|
Établir et tenir à jour un dossier contenant ou identifiant des documents qui définissent le respect des exigences de la norme ISO 13485 et des exigences réglementaires applicables | Cf. § 4.2.5. Lien avec le marquage CE du dispositif médical | |
|
37
|
4.2.3 a
|
Inclure dans ce dossier la description générale du dispositif médical, l'utilisation prévue, l'étiquetage | Y compris des instructions d'utilisation. Plus d'informations dans l'ISO 15223 (Dispositifs médicaux - Symboles à utiliser avec les étiquettes, l'étiquetage et les informations à fournir relatifs aux dispositifs médicaux - Partie 1: Exigences générales) | |
|
38
|
4.2.3 b
|
Inclure dans ce dossier les spécifications du produit | Cf. § 7.2.1 | |
|
39
|
4.2.3 c
|
Inclure dans ce dossier les spécifications de fabrication, emballage, stockage, manipulation et distribution | Cf. § 7.2.1 | |
|
40
|
4.2.3 d
|
Inclure dans ce dossier la procédure de mesurage et surveillance | Cf. § 7.6 | |
|
41
|
4.2.3 e
|
Inclure dans ce dossier les exigences d'installation | Selon le cas, cf. § 7.5.3 | |
|
42
|
4.2.3 f
|
Inclure dans ce dossier la procédure de service | Selon le cas, cf. § 7.5.4 | |
|
|
4.2.4 |
Maîtrise des documents
|
|
|
|
43
|
4.2.4
|
Maîtriser les documents requis du SMQ | La procédure définit comment avant de commencer à utiliser un document, celui-ci est approuvé (vérifié, validé) par une personne avec des responsabilités et autorités définies | |
|
44
|
4.2.4
|
Maîtriser les enregistrements | Cf. § 4.2.5. Les enregistrements sont des documents particuliers | |
|
45
|
4.2.4 a
|
Passer en revue et approuver les documents | Quant à leur adéquation avant diffusion | |
| 46 |
4.2.4 b
|
Passer en revue et approuver de nouveau les documents | Mettre à jour, si nécessaire | |
| 47 | 4.2.4 c | Assurer l'identification des modifications et de l'état de la version en vigueur | La gestion des modifications et indices (versions) des documents est réalisée par une personne avec des responsabilités et autorités établies | |
|
48
|
4.2.4 d
|
Assurer la disponibilité sur les lieux d'utilisation des versions pertinentes des documents applicables | "Le bon document, au bon endroit, au bon moment" et avec la bonne version | |
| 49 |
4.2.4 e
|
Assurer que les documents restent lisibles et facilement identifiables | Chaque document est clair, simple à comprendre, facile à catégoriser y compris les fichiers électroniques | |
| 50 | 4.2.4 f | Assurer que les documents d'origine extérieure sont identifiés et que leur diffusion est maîtrisée | Les documents externes déterminés par l'entreprise comme nécessaires pour la planification et le fonctionnement du SMQ,(normes, cahiers des charges, spécifications) sont identifiés et maîtrisés (liste, emplacement, version). Méthode de codification des documents | |
|
51
|
4.2.4 g
|
Prévenir la détérioration et la perte de documents | Y compris les fichiers électroniques (sauvegardes régulières) | |
| 52 |
4.2.4 h
|
Empêcher l'utilisation de documents périmés | Les documents périmés (obsolètes) sont identifiés, conservés, archivés, enfermés ou détruits de sorte que l’on ne puisse pas les utiliser normalement | |
| 53 | 4.2.4 | S'assurer que les modifications apportées sont revues et approuvées par qui de droit | Soit par l'autorité d'approbation d'origine, soit par une personne disposant d'informations pertinentes | |
| 54 | 4.2.4 | Définir la période de conservation des documents périmés | Cf. § 4.2.5 | |
| 55 | 4.2.4 | Assurer une période de conservation des documents au moins égale à la durée de vie du dispositif médical mais non inférieure à celle spécifiée par les exigences réglementaires applicables | Liste des procédures documentées obligatoires :
|
|
|
|
4.2.5 |
Maîtrise des enregistrements
|
|
|
|
56
|
4.2.5
|
Conserver les enregistrements comme preuve du respect des exigences | "Les paroles s'envolent, les écrits restent. Proverbe latin". Les enregistrements sont renseignés quotidiennement (sans retard). Sans eux c’est difficile (voire impossible) d'apporter la preuve du respect des exigences du SMQ |
|
| 57 | 4.2.5 | Apporter la preuve de l'efficacité du SMQ | En conservant les enregistrements | |
|
58
|
4.2.5
|
Documenter la procédure maîtrise des enregistrements | Cf. § 4.2.4. La procédure pour les enregistrements répond aux questions qui, quand, comment, dans quelles conditions identifier, stocker, appliquer des mesures pour la sécurité et l'intégrité, récupérer, conserver et éliminer les enregistrements | |
| 59 | 4.2.5 | Définir et appliquer des méthodes de protection des informations confidentielles de santé | Conformément aux exigences réglementaires applicables | |
| 60 | 4.2.5 | Maintenir les enregistrements lisibles, faciles à identifier et accessibles | Chaque enregistrement est clair, simple à comprendre, facile à catégoriser y compris les fichiers électroniques | |
| 61 | 4.2.5 | Maintenir les modifications aux enregistrements identifiables | Règles à appliquer (par exemple nom, signature, date, justification) | |
| 62 | 4.2.5 | Conserver les enregistrements au moins pendant le cycle de vie du dispositif médical | Conformément aux exigences de l'entreprise ou des exigences réglementaires applicables mais plus de 2 ans après la distribution du dispositif médical. Liste des enregistrements obligatoires :
|
|
|
|
Responsabilité de la direction
|
|||
|
|
Engagement de la direction
|
|||
|
63
|
5.1
|
Apporter la preuve de l'engagement de la direction au développement, la mise en œuvre et au maintien de l'efficacité du SMQ | "Un escalier se balaie en commençant par le haut. Proverbe roumain" | |
| 64 | 5.1 a | Communiquer l'importance à satisfaire aux exigences des clients | Et des exigences réglementaires applicables | |
|
65
|
5.1 b
|
Établir la politique qualité | Définir la politique qualité est une implication incontournable, directe et documentée de la direction pour mettre en place et maintenir l’efficacité du SMQ | |
|
66
|
5.1 c
|
S'assurer que les objectifs qualité sont établis | Quantifier dans chaque service des objectifs qualité cohérents avec la politique qualité et les exigences clients | |
|
67
|
5.1 d
|
Mener les revues de direction | Cf. § 5.6. Garder les comptes rendus des revues de direction, qui sont la preuve que le SMQ est pertinent, efficace et en permanente amélioration | |
|
68
|
5.1 e
|
Assurer la disponibilité des ressources | Cf. § 6.1. La direction fournit les ressources nécessaires pour réaliser la politique qualité et atteindre les objectifs fixés |
|
|
|
Ecoute client
|
|
||
|
69
|
5.2
|
Déterminer et respecter les exigences des clients | Et des exigences réglementaires applicables. Cf. §§ 7.2.1 et 8.2.1 | |
|
|
Politique qualité
|
|||
|
70
|
5.3 a
|
Adapter la politique qualité à la finalité de l'entreprise | La politique qualité est cohérente avec la satisfaction du client et l’amélioration continue du SMQ | |
|
71
|
5.3 b
|
Inclure l'engagement à satisfaire aux exigences et maintenir l'efficacité du SMQ | Engagement sans équivoque de la direction | |
|
72
|
5.3 c
|
Fournir le cadre pour établir les objectifs qualité | Et passer en revue les objectifs qualité. Cf. § 5.6. La revue de direction est le cadre par excellence pour cette exigence | |
|
73
|
5.3 d
|
Communiquer et expliquer la politique qualité | Cf. § 5.5.3 | |
|
74
|
5.3 e
|
Passer en revue la politique qualité | Cf. § 5.6. La politique qualité évolue en permanence. C’est l'un des objectifs de la revue de direction |
|
|
|
5.4
|
Planification
|
|
|
|
|
Objectifs qualité
|
|||
| 75 |
5.4.1
|
Établir les objectifs qualité aux niveaux appropriés de l'entreprise | Y compris ceux nécessaires à la satisfaction aux exigences réglementaires applicables et ceux du produit. Cf. § 7.1 | |
| 76 | 5.4.1 | Assurer des objectifs qualités mesurables | Cf. § 7.1 | |
|
77
|
5.4.1
|
Assurer des objectifs qualité cohérents avec la politique qualité | Les objectifs qualité sont chiffrés, traduits (déclinés) en indicateurs et suivis régulièrement (tableaux de bord). Un critère de mesurabilité peut être "Oui/Non" | |
|
|
Planification du système de management de la qualité
|
|||
|
78
|
5.4.2 a
|
Respecter l'approche processus lors de la planification du SMQ | Et les objectifs qualité. Cf. § 4.1 | |
|
79
|
5.4.2 b
|
Tenir à jour la cohérence du SMQ pendant la planification et mise en œuvre des modifications | Cf. § 7.3.9. Attention particulière sur la maîtrise des modifications et leurs conséquences sur la performance du SMQ | |
|
|
Responsabilité, autorité et communication
|
|
||
|
|
Responsabilité et autorité
|
|
||
|
80
|
5.5.1
|
Définir, documenter et communiquer les responsabilités et autorités | Cf. § 4.2.5. "La responsabilité ne peut pas être partagée. Robert Heilein". Des descriptions de fonction claires et disponibles en interne (aussi organigramme, matrice de compétence) | |
| 81 | 5.5.1 | Documenter les liens entre les personnes chargées de gérer, de réaliser et d'évaluer un travail ayant une incidence sur la qualité | C'est un engagement que la direction valide quand ce n'est pas elle-même qui définit ces liens | |
| 82 | 5.5.1 | Assurer l'autonomie et l'autorité nécessaires pour réaliser ces tâches | Cf. § 8.2.1 | |
|
|
Représentant de la direction
|
|||
|
83
|
5.5.2
|
Nommer le représentant de la direction | Il est membre de l’encadrement et ne fait pas partie obligatoirement du service qualité | |
|
84
|
5.5.2 a
|
Documenter les processus nécessaires au SMQ par le représentant de la direction | Cf. §§ 7.2, 7.5 et 4.2.4 | |
| 85 | 5.5.2 b | Rendre compte à la direction du fonctionnement du SMQ par le représentant de la direction | Et des besoins en amélioration, cf. § 8.5 | |
| 86 | 5.2.2 c | Assurer la sensibilisation du personnel aux exigences réglementaires applicables et du SMQ par le représentant de la direction | Inclure dans la description de fonction du représentant de la direction les tâches liées à la sensibilisation du personnel aux différentes exigences | |
|
|
Communication interne
|
|||
|
87
|
5.5.3
|
Établir les processus de communication appropriés | Attention particulière au retour d'information du personnel (enquêtes, boîte à suggestions) | |
| 88 | 5.5.3 | Communiquer sur l'efficacité du SMQ | C'est un engagement de la direction | |
|
|
Revue de direction
|
|
||
|
|
Généralités
|
|
||
|
89
|
5.6.1
|
Documenter la procédure revue de direction | Cf. § 4.2.4. "Aucun système n'est parfait". D’habitude une ou deux fois par an, passer en revue la totalité du SMQ pour vérifier l’atteinte des objectifs qualité | |
|
90
|
5.6.1
|
Passer en revue le SMQ à intervalles planifiés | Pour s'assurer qu'il est toujours pertinent, adéquat et efficace. Passer en revue les opportunités (occasions, conditions, possibilités, circonstances) d’amélioration continue du SMQ | |
|
91
|
5.6.1
|
Évaluer les opportunités d'amélioration | Passer en revue les opportunités (occasions, conditions, possibilités, circonstances) d’amélioration continue du SMQ | |
|
92
|
5.6.1
|
Évaluer le besoin de modifier le SMQ | Y compris la politique qualité et les objectifs qualité. Passer en revue les opportunités (occasions, conditions, possibilités, circonstances) d’amélioration continue du SMQ | |
|
93
|
5.6.1
|
Conserver les enregistrements de la revue de direction |
Cf. § 4.2.5 | |
|
|
5.6.2
|
Éléments d’entrée de la revue
|
|
|
|
94
|
5.6.2 a
|
Inclure les informations sur les retours d'information | Si c'est le premier élément, c'est souvent le plus important | |
|
95
|
5.6.2 b
|
Inclure les informations sur les réclamations | "Aimez vos clients plus que vos produits". Toutes les données de satisfaction et de non satisfaction des clients sont une source importante d’informations pour trouver des opportunités d'amélioration du SMQ | |
|
96
|
5.6.2 c
|
Inclure les informations sur le signalement aux autorités réglementaires | Cf. § 8.2.3 | |
| 97 | 5.6.2 d | Inclure les informations sur les audits | Les rapports d’audits internes et leurs propositions sont une source importante d’informations pour améliorer le SMQ | |
| 98 |
5.6.2 e
|
Inclure les informations sur les processus de surveillance et de mesurage | Résultats de l'atteinte des objectifs qualité et analyse des données liées aux dysfonctionnements (non-conformités) des processus | |
| 99 |
5.6.2 f
|
Inclure les informations sur la surveillance et le mesurage du produit | Résultats et tendances | |
|
100
|
5.6.2 g
|
Inclure les informations sur les actions correctives | Résultats des actions, leur suivi, les améliorations obtenues | |
|
101
|
5.6.2 h
|
Inclure les informations sur les actions préventives | Résultats des actions, leur suivi, les améliorations obtenues | |
|
102
|
5.6.2 i
|
Inclure les informations sur les actions issues des revues de directions précédentes | Résultats des décisions prises pendant la dernière revue de direction et leur suivi | |
|
103
|
5.6.2 j
|
Inclure les informations sur les modifications pouvant affecter le SMQ | Prendre en compte, évaluer et analyser toute modification pouvant avoir un impact sur le SMQ (nouveaux produits et processus, nouveaux clients, nouvelles fonctions et responsabilités) | |
|
104
|
5.6.2 k
|
Inclure les informations sur les recommandations d'amélioration | Suggestions, avis, opinions, propositions venant de l’ensemble du personnel ou des parties prenantes externes | |
|
105
|
5.6.2 l
|
Inclure les informations sur les exigences réglementaires nouvelles ou révisées | Veille réglementaire (évolutions des exigences réglementaires applicables) | |
|
|
Éléments de sortie de la revue
|
|
||
|
106
|
5.6.3
|
Enregistrer les éléments de sortie de la revue de direction | Cf. § 4.2.5. Après analyse et revue de tous les éléments d'entrée, formaliser les décisions | |
|
107
|
5.6.3 a
|
Décider les actions relatives à l'amélioration du SMQ | Afin de maintenir la pertinence, l'adéquation et l'efficacité du SMQ | |
|
108
|
5.6.3 b
|
Décider les actions relatives à l'amélioration du produit | Par rapport aux exigences du client | |
|
109
|
5.6.3 c
|
Décider les actions relatives aux modifications nécessaires | Pour répondre aux exigences réglementaires applicables nouvelles ou révisées | |
|
110
|
5.6.3 d
|
Décider les actions relatives aux besoins en ressources | Afin de répondre aux besoins nécessaires en personnel et ressources matérielles | |
|
|
Management des ressources
|
Planifier (Plan)
|
||
|
|
Mise à disposition des ressources
|
|
||
|
111
|
6.1 a
|
Déterminer et fournir les ressources pour mettre en œuvre et maintenir l'efficacité du SMQ | Identifier et assurer les besoins présents et futurs en ressources : - personnel (compétences) - infrastructures (bâtiments, espaces de travail, installations) - équipements (logiciels, matériels) des processus - services supports (logistique, communication) |
|
|
112
|
6.1 b
|
Déterminer et fournir les ressources pour satisfaire aux exigences réglementaires et du client applicables | "La bonne personne, au bon endroit, au bon moment" | |
|
Personnel
|
||||
| 113 |
6.2
|
Assurer les compétences du personnel effectuant un travail ayant une incidence sur la qualité du produit | Les compétences sont fondées sur la formation initiale et professionnelle, le savoir-faire et l'expérience | |
| 114 |
6.2
|
Documenter le processus nécessaire pour définir les compétences, dispenser la formation et assurer la sensibilisation du personnel | Cf. § 4.2.4. "La qualité c'est l'affaire de tous". Sensibiliser le personnel sur le lien entre la formation et la responsabilité individuelle pour atteindre les objectifs qualité | |
| 115 |
6.2 a
|
Déterminer les compétences nécessaires | Pour le personnel effectuant un travail ayant une incidence sur la qualité du produit | |
| 116 |
6.2 b
|
Pourvoir à la formation | Ou identifier d'autres actions à entreprendre pour acquérir ou entretenir les compétences nécessaires. | |
| 117 |
6.2 c
|
Vérifier l'efficacité de la formation | La méthode est proportionnée au niveau de risque associé (impacts sur la sécurité et la conformité réglementaire des dispositifs médicaux) | |
| 118 |
6.2 d
|
Assurer que le personnel est conscient de la pertinence et de l'importance de leurs activités | Et de la manière dont ils contribuent à la réalisation des objectifs qualité | |
| 119 |
6.2 e
|
Conserver les enregistrements concernant la formation initiale et professionnelle, le savoir-faire et l'expérience | Cf. § 4.2.5 | |
|
|
Infrastructures
|
|
||
| 120 | 6.3 | Documenter les exigences relatives aux infrastructures nécessaires | Cf. § 4.2.4. Pour satisfaire aux exigences du produit, empêcher leur mélange et assurer leur manutention ordonnée | |
|
121
|
6.3
|
Documenter les exigences des activités de maintenance y compris leur périodicité | Cf. § 4.2.4. Surtout lorsque ces activités ou leur absence peuvent avoir une incidence sur la qualité du produit | |
|
122
|
6.3
|
Appliquer ces exigences aux équipements de maintenance | Équipements utilisés pour la production, la maîtrise de l'environnement de travail, la surveillance et le mesurage | |
|
123
|
6.3
|
Conserver les enregistrements des activités de maintenance | Cf. § 4.2.5 | |
|
|
Environnement de travail et maîtrise de la contamination
|
|||
|
|
Environnement de travail
|
|||
|
124
|
6.4.1
|
Documenter les exigences relatives à l'environnement de travail nécessaire | Cf. § 4.2.4. Tout ce qui peut avoir un impact sur la conformité du produit (motivation, organisation du travail, ergonomie des postes de travail, éclairage, hygiène, température, sécurité) | |
| 125 | 6.4.1 | Documenter les exigences relatives à l'environnement de travail et la procédure pour le surveiller et le maîtriser | Cf. § 4.2.4. Surtout si l'environnement de travail peut avoir une incidence négative sur la qualité du produit. Cf. § 7.5.1 | |
| 126 | 6.4.1 a | Documenter les exigences en matière de santé, propreté et habillement du personnel | Cf. § 4.2.4. Dans les cas de contact du personnel ou l'environnement de travail avec le produit. Cf. § 7.5.2 | |
| 127 | 6.4.1 b | S'assurer que les personnes qui doivent travailler dans des conditions d'environnement particulières sont compétentes | Ou surveillées par une personne compétente. Cf. § 6.2. Plus d'informations dans l'ISO 14644 (Salles propres et environnements maîtrisés apparentés - Partie 1: Classification de la propreté particulaire de l'air) et l'ISO 14698 (Salles propres et environnements maîtrisés apparentés - Maîtrise de la bio contamination - Partie 1: Principes généraux et méthodes) | |
|
|
6.4.2
|
Maîtrise de la contamination
|
||
|
128
|
6.4.2
|
Planifier et documenter les dispositions pour la maîtrise du produit contaminé ou potentiellement contaminé | Cf. § 4.2.4. Afin d'éviter la contamination de l'environnement de travail, du personnel ou du produit. Cf. § 7.5.5 | |
|
129
|
6.4.2
|
Documenter les exigences pour la maîtrise de la contamination (particulaire et microbiologique) | Cf. § 4.2.4. Pour les dispositifs médicaux stériles | |
|
130
|
6.4.2
|
Maintenir la propreté requise en assemblage et conditionnement | Pour les dispositifs médicaux stériles lister les mesures nécessaires mises en place | |
|
|
Réalisation du produit
|
Dérouler (Do)
|
||
|
|
Planification de la réalisation du produit
|
|
||
|
131
|
7.1
|
Planifier les processus de réalisation du produit | Cf. § 4.2.5. Ce sont tous les processus qui répondent aux besoins et attentes des clients (de la demande de devis jusqu’au service après-vente). Une cartographie des processus peut éclaircir l’image globale de la réalisation du produit | |
| 132 | 7.1 | Développer les processus de réalisation du produit | Prendre en compte les infrastructures et l'environnement de travail, cf. §§ 6.3 et 6.4 | |
| 133 |
7.1
|
Planifier la réalisation du produit en cohérence avec les exigences relatives aux autres processus du SMQ | Cf. § 4.1. Prendre en compte autant que possible la prévention | |
| 134 |
7.1
|
Documenter les processus de gestion des risques tout au long des processus de réalisation du produit | Cf. § 4.2.4. Plus d'informations dans l'ISO 14971 (Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux) | |
| 135 | 7.1 | Conserver les enregistrements des activités de gestion des risques | Cf. § 4.2.5 | |
| 136 |
7.1 a
|
Déterminer les objectifs qualité et les exigences du produit | Établir et mettre en place les processus et la documentation pour chaque produit | |
| 137 | 7.1 b | Déterminer le besoin d'établir des processus et des documents | Cf. § 4.2.4 | |
| 138 | 7.1 b | Déterminer comment fournir les ressources spécifiques au produit | Y compris les infrastructures et l'environnement de travail | |
| 139 |
7.1 c
|
Déterminer les activités de vérification, validation, surveillance, mesure, inspection et essai, stockage, distribution | Et les activités de traçabilité spécifiques au produit | |
| 140 | 7.1 c | Déterminer les critères d'acceptation du produit | Et les appliquer sans exception | |
| 141 |
7.1 d
|
Déterminer les enregistrements spécifiques au processus | Cf. § 4.2.5. Afin d'apporter des preuves de satisfaction aux exigences | |
| 142 | 7.1 d | Déterminer les enregistrements spécifiques au produit | Cf. § 4.2.5. Afin d'apporter des preuves de satisfaction aux exigences | |
| 143 |
7.1
|
Assurer que les éléments de sortie de la planification sont sous une forme adéquate | Cf. § 4.2.5. Papier ou électronique, facilement lisible et identifiable | |
|
|
Produits clients
|
|
||
|
|
Détermination des exigences relatives au produit
|
|
||
|
144
|
7.2.1 a
|
Déterminer les exigences spécifiées par le client y compris celles relatives à la livraison et après livraison | "La seule mesure de la qualité est la satisfaction du client". Identifier et appliquer les besoins et attentes clients en exigences internes du produit (réalisation, livraison et après livraison) | |
|
145
|
7.2.1 b
|
Déterminer les exigences pour l'usage prévu | Identifier et appliquer les besoins et attentes implicites clients (garantie à vie, fiabilité exemplaire, maintenance simple) | |
|
146
|
7.2.1 c
|
Déterminer les exigences réglementaires applicables | Identifier toutes les exigences applicables au produit (y compris recyclage et élimination) et mettre en place une veille réglementaire | |
|
147
|
7.2.1 d
|
Déterminer toute formation de l'utilisateur nécessaire pour assurer les performances spécifiées | Et l'utilisation sûre du dispositif médical | |
|
148
|
7.2.1 e
|
Déterminer toute exigence complémentaire interne | Comme contraintes, règlement intérieur, confidentialité, santé et sécurité au travail, hygiène | |
|
|
7.2.2
|
Revue des exigences relatives au produit
|
||
|
149
|
7.2.2
|
Passer en revue les exigences relatives au produit | Passer en revue en amont les exigences du produit (faisabilité, rentabilité) | |
|
150
|
7.2.2
|
Mener la revue avant de s’engager à livrer le produit au client | Passer en revue les devis, les amendements, l'acceptation des commandes, les modifications | |
| 151 | 7.2.2 a | S'assurer que les exigences sont définies et documentées | Cf. § 4.2.4 | |
|
152
|
7.2.2 b
|
S'assurer que les écarts entre les exigences d'un contrat ou d'une commande et celles précédemment exprimées ont été résolus | Avant d'accepter le contrat ou la commande | |
|
153
|
7.2.2 c
|
S'assurer que les exigences réglementaires applicables sont satisfaites | Les exigences sont définies et approuvées | |
|
154
|
7.2.2 d
|
S'assurer que toute formation de l'utilisateur est disponible ou planifiée de l'être | Cf. § 7.2.1 | |
|
155
|
7.2.2 e
|
S'assurer que l'entreprise est apte à satisfaire aux exigences | Les exigences sont définies et approuvées | |
|
156
|
7.2.2
|
Conserver les enregistrements des résultats de la revue et des actions entreprises | Cf. § 4.2.5 | |
|
157
|
7.2.2
|
Lorsque les exigences du client ne sont pas documentées, confirmez les avant de les accepter | En les envoyant au client avec accusé de réception (par la poste ou par courriel) | |
|
158
|
7.2.2
|
Lorsque les exigences relatives au produit sont modifiées, s'assurer que les documents correspondants sont amendés | Et que le personnel concerné est informé | |
|
|
Communication
|
|
||
|
159
|
7.2.3 a
|
Planifier et documenter les dispositions pour communiquer avec les clients les informations relatives au produit | Cf. § 4.2.5. "Les bonnes nouvelles marchent et les mauvaises courent. Proverbe suédois" | |
| 160 | 7.2.3 b | Planifier et documenter les dispositions pour communiquer avec les clients le traitement des consultations, contrats, commandes et leurs avenants | Cf. § 4.2.5. Mettre en place des méthodes de communication efficaces avec le client | |
|
161
|
7.2.3 c
|
Planifier et documenter les dispositions pour communiquer avec les clients les retours d'information des clients | Cf. § 4.2.5. Y compris les réclamations. Cf. § 8.2.1 | |
| 162 | 7.2.3 d | Planifier et documenter les dispositions pour communiquer avec les clients les fiches d'avertissement | Cf. §§ 8.3.3 et 4.2.4 | |
| 163 | 7.2.3 | Communiquer avec les autorités réglementaires conformément aux exigences réglementaires applicables | Cf. § 8.2.3 | |
|
|
Conception et développement
|
|
||
|
|
Généralités
|
|
||
| 164 | 7.3.1 | Documenter des procédures pour la conception et le développement | Cf. § 4.2.4. "Je n'ai pas échoué. J'ai juste trouvé 10 000 moyens qui ne fonctionnent pas. Thomas Edison" | |
|
|
7.3.2
|
Planification de la conception et du développement
|
|
|
|
165
|
7.3.2
|
Planifier et maîtriser la la conception et le développement du produit | Identifier et formaliser les relations entre tous les acteurs de la conception et du développement du produit (réunions et revues aux étapes clés) | |
| 166 | 7.3.2 | Conserver et tenir à jour les documents de la planification autant que nécessaire | Au cours du déroulement de la conception et du développement | |
|
167
|
7.3.2 a
|
Documenter les étapes de la conception et du développement | Cf. § 4.2.4 | |
|
168
|
7.3.2 b
|
Documenter les revues nécessaires à chaque étape de la conception et du développement | Cf. § 4.2.4 | |
|
169
|
7.3.2 c
|
Documenter les activités de vérification, validation et de transfert de conception appropriées à chaque étape de la conception et du développement | Cf. § 4.2.4 | |
| 170 | 7.3.2 d | Documenter les responsabilités et autorités pour la conception et le développement | Cf. § 4.2.4 | |
|
171
|
7.3.2 e
|
Documenter les méthodes permettant d'assurer la traçabilité des éléments de sortie | Cf. § 4.2.4. Par rapport aux éléments d'entrée de la conception et du développement | |
|
172
|
7.3.2 f
|
Documenter les ressources nécessaires | Cf. § 4.2.4. Y compris les compétences du personnel | |
|
|
Eléments d’entrée de la conception et du développement
|
|
||
|
173
|
7.3.3
|
Déterminer les éléments d'entrée relatifs aux exigences du produit | Caractéristiques du produits et spécifications techniques (emballage et autres) | |
|
174
|
7.3.3
|
Conserver les enregistrements des éléments d'entrée | Cf. § 4.2.5 | |
|
175
|
7.3.3 a
|
Inclure les exigences d'aptitude à l'utilisation | Caractéristiques fonctionnelles, de performance et de sécurité | |
|
176
|
7.3.3 b
|
Inclure les exigences réglementaires applicables | Et les normes spécifiques | |
|
177
|
7.3.3 c
|
Inclure des éléments de sortie de la gestion des risques | Cf. § 7.1 | |
|
178
|
7.3.3 d
|
Inclure des informations de conceptions similaires précédentes | Pourquoi réinventer la roue ? | |
|
179
|
7.3.3 e
|
Inclure d'autres exigences essentielles pour la conception et le développement du produit et des processus | Analyse des concurrents, étude du marché | |
|
180
|
7.3.3
|
Passer en revue l'adéquation des éléments d'entrée | Par rapport à l'usage prévu | |
|
181
|
7.3.3
|
Approuver les éléments d'entrée | Par une personne avec des responsabilité et autorités établies. Cf. § 5.5.1 | |
|
182
|
7.3.3
|
S'assurer que les exigences sont complètes, non ambiguës, vérifiables, valides et sans conflits | Plus d'informations dans l'IEC 62366-1 (Dispositifs médicaux - Partie 1: Application de l'ingénierie de l'aptitude à l'utilisation aux dispositifs médicaux) | |
|
|
Eléments de sortie de la conception et du développement
|
|||
|
183
|
7.3.4 a
|
S'assurer que les éléments de sortie de la conception et du développement satisfont aux exigences d'entrée | Si ce n'est pas le cas, recommencer certaines étapes | |
| 184 | 7.3.4 b | S'assurer que les éléments de sortie de la conception et du développement fournissent les informations appropriées pour les achats, la production et la prestation de service | La précision de ces informations est essentielle | |
| 185 |
7.3.4 c
|
S'assurer que les éléments de sortie de la conception et du développement contiennent ou font référence aux critères d'acceptation | Les exigences (restrictions ou recommandations) liées au transport, à l'emballage, aux étiquettes, aux notices d'utilisation, à la date limite d'utilisation, à la traçabilité, aux composants utilisés | |
| 186 |
7.3.4 d
|
S'assurer que les éléments de sortie de la conception et du développement spécifient les caractéristiques du produit essentielles pour son utilisation correcte et en toute sécurité | Les résultats des revues de conception de produits similaires anciens (tests, fiabilité, faisabilité) et retours d'information des usagers (service après-vente, recommandations, suggestions) | |
| 187 |
7.3.4
|
S'assurer que les éléments de sortie de la conception et du développement sont fournis sous une forme permettant leur vérification par rapport aux éléments d'entrée | Exigence inclue dans la procédure de conception et développement, cf. § 7.3.1 | |
| 188 | 7.3.4 | S'assurer que les éléments de sortie de la conception et du développement sont approuvés avant leur mise à disposition | Exigence inclue dans la procédure de conception et développement, cf. § 7.3.1 | |
|
189
|
7.3.4
|
Conserver les enregistrements des éléments de sortie de la conception et du développement | Cf.§ 4.2.5 | |
|
|
7.3.5
|
Revue de la conception et du développement
|
|
|
|
190
|
7.3.5
|
Mener des revues méthodiques de la conception et du développement aux étapes appropriées | Conformément aux dispositions planifiées et documentées | |
|
191
|
7.3.5 a
|
Évaluer l'aptitude des résultats à satisfaire aux exigences | Cf. § 7.3.3 | |
|
192
|
7.3.5 b
|
Proposer les actions nécessaires | Après avoir identifié les problèmes | |
|
193
|
7.3.5
|
S'assurer que les représentants des fonctions concernées par les étapes de la revue participent à ces revues | Et d'autres personnes spécialisées | |
|
194
|
7.3.5
|
Conserver les enregistrements des revues et des actions nécessaires | Cf. § 4.2.5. Ces enregistrements incluent l'identification de la conception objet de la revue, les participants impliqués et la date de la revue. Exigence inclue dans la procédure de conception et développement, cf. § 7.3.1 | |
|
|
7.3.6
|
Vérification de la conception et du développement
|
||
|
195
|
7.3.6
|
Réaliser la vérification de la conception et du développement conformément aux dispositions planifiées et documentées | Afin de s'assurer que les éléments de sortie satisfont aux exigences des éléments d'entrée | |
| 196 | 7.3.6 | Documenter les plans de vérification | Cf. § 4.2.4. Ces plans incluent les méthodes, les critères d'acceptation, les techniques statistiques avec la taille d'échantillonnage | |
| 197 |
7.3.6
|
Inclure dans la vérification la confirmation que les éléments de sortie de la conception satisfont les éléments d'entrée de la conception pendant le raccord ou en interface | Lorsque l'usage prévu exige que le dispositif médical est raccordé ou est en interface avec d'autres dispositifs | |
| 198 |
7.3.6
|
Conserver les enregistrements des résultats et conclusions de la vérification et des actions nécessaires | Cf. §§ 4.2.4 et 4.2.5 | |
|
|
7.3.7
|
Validation de la conception et du développement
|
|
|
|
199
|
7.3.7
|
Réaliser la validation de la conception et du développement conformément aux dispositions planifiées et documentées | Afin de s'assurer que le produit résultant est apte à satisfaire aux exigences pour l'application spécifiée ou l'usage prévu | |
|
200
|
7.3.7
|
Documenter les plans de validation | Cf. § 4.2.4. Ces plans incluent les méthodes, les critères d'acceptation, les techniques statistiques avec la taille d'échantillonnage | |
|
201
|
7.3.7
|
Conduire la validation de la conception sur un produit représentatif | Le produit représentatif inclut des échantillons initiaux de production, des lots ou leurs équivalents | |
|
202
|
7.3.7
|
Conserver la justification du choix du produit utilisé pour validation | Cf. § 4.2.5 | |
|
203
|
7.3.7
|
Réaliser des évaluations cliniques ou évaluations du fonctionnement du dispositif médical conformément aux exigences réglementaires applicables dans le cadre de la validation de la conception et du développement | La mise à disposition du dispositif médical à des fins d'évaluations cliniques ou évaluations du fonctionnement n'est pas considérée comme livraison effective pour le client | |
|
204
|
7.3.7
|
Inclure dans la validation la confirmation que les exigences pour l'application spécifiée ou l'usage prévu ont été satisfaits pendant le raccord ou en interface | Lorsque l'usage prévu exige que le dispositif médical est raccordé ou est en interface avec d'autres dispositifs | |
|
205
|
7.3.7
|
Réaliser la validation avant la libération pour utilisation du produit par le client | Cf. §§ 7.5.6 et 7.5.7 | |
|
206
|
7.3.7
|
Conserver les enregistrements des résultats et conclusions de la validation et des actions nécessaires | Cf. §§ 4.2.4 et 4.2.5 | |
|
|
7.3.8
|
Transfert de la conception et du développement
|
||
|
207
|
7.3.8
|
Documenter des procédures pour le transfert des éléments de sortie de la conception et du développement à la fabrication | Cf. § 4.2.4. Identifier, formaliser et garder les informations nécessaires pour lancer la conception et le développement du produit | |
| 208 | 7.3.8 | S'assurer que ces procédures permettent de vérifier que les éléments de sortie de la conception et du développement sont adaptés à la fabrication | Avant de devenir des spécifications finales de production | |
| 209 |
7.3.8
|
S'assurer que ces procédures permettent de vérifier que les moyens de production peuvent satisfaire aux exigences du produit | Cf. § 7.2.1 | |
| 210 |
7.3.8
|
Enregistrer les résultats et conclusions du transfert | Cf. § 4.2.5 | |
|
|
Maîtrise des modifications de la conception et du développement
|
|
||
|
211
|
7.3.9
|
Documenter des procédures pour maîtriser les modifications de la conception et du développement | Cf. § 4.2.4. Identifier et formaliser les relations entre tous les acteurs de la conception et du développement du produit (réunions et revues aux étapes clés) | |
|
212
|
7.3.9
|
Déterminer la conséquence de la modification | Sur la fonction, les performances, l'aptitude à l'utilisation, la sécurité du produit, les exigences réglementaires applicables et l'usage prévu du dispositif médical | |
|
213
|
7.3.9
|
Identifier les modifications de la conception et du développement | Avec des règles (version du document) et méthodes (encre rouge, signature, date) | |
|
214
|
7.3.9 a
|
Passer en revue les modifications | Avant leur mise en œuvre | |
|
215
|
7.3.9 b
|
Vérifier les modifications | Avant leur mise en œuvre | |
|
216
|
7.3.9 c
|
Valider les modifications | Le cas échéant, avant leur mise en œuvre | |
|
217
|
7.3.9
|
Approuver les modifications | Avant leur mise en œuvre | |
|
218
|
7.3.9
|
Inclure pendant la revue des modifications de la conception et du développement l'évaluation de l'incidence des modifications | Sur les composants du produit, sur le produit en cours de livraison ou déjà livré, sur les éléments d'entrée ou de sortie de la gestion des risques et sur les processus de réalisation du produit | |
|
219
|
7.3.9
|
Conserver les enregistrements des modifications, leur revue et les actions nécessaires | Cf. § 4.2.5 | |
|
|
7.3.10
|
Dossiers de la conception et du développement
|
||
|
220
|
7.3.10
|
Tenir à jour un dossier de conception et de développement par type ou famille de dispositif médical | Cf. § 4.2.4 | |
| 221 | 7.3.10 | Inclure ou référencer dans ce dossier des enregistrements pour démontrer la satisfaction aux exigences de la conception et du développement | Cf. §§ 4.2.5 et 7.3.4 | |
| 222 |
7.3.10
|
Inclure ou référencer dans ce dossier des enregistrements des modifications de la conception et du développement | Cf. §§ 4.2.5 et 7.3.9 | |
|
|
Achats
|
|
||
|
|
Processus d'achat
|
|
||
| 223 | 7.4.1 | Documenter des procédures pour s'assurer que le produit acheté est conforme aux informations d'achat spécifiés | Cf. § 4.2.4. Les informations sont des exigences internes | |
|
224
|
7.4.1
|
Établir les critères d'évaluation et de sélection des fournisseurs | Le processus d'achat inclut les critères d'évaluation permanente (mensuelle ou trimestrielle) des fournisseurs (% de produits achetés non-conformes détectés en inspection entrée, en production et en post-production) | |
|
225
|
7.4.1 a
|
S'assurer que les critères sont fondés sur la capacité du fournisseur à fournir un produit qui satisfait les exigences de l'entreprise | Cf. § 7.2.1 | |
|
226
|
7.4.1 b
|
S'assurer que les critères sont fondés sur les performances du fournisseur | Produit conforme, respect du coût et des délais | |
|
227
|
7.4.1 c
|
S'assurer que les critères sont fondés sur l'incidence du produit acheté sur la qualité du dispositif médical | "Acheter de la qualité, c'est choisir de ne pleurer qu'une seule fois. Proverbe anglais" | |
|
228
|
7.4.1 d
|
S'assurer que les critères sont proportionnés au risque associé au dispositif médical | Cf. § 7.1 | |
|
229
|
7.4.1
|
Planifier la surveillance et la réévaluation des fournisseurs | Inclure dans la liste des fournisseurs l'historique des évaluations | |
|
230
|
7.4.1
|
Surveiller les performances des fournisseurs | Par rapport au respect des exigences du produit acheté. Les activités de surveillance sont décrites dans la procédure d'achat | |
|
231
|
7.4.1
|
Fournir comme élément d'entrée du processus de réévaluation des fournisseurs les résultats de la surveillance | Utiliser les enregistrements de la vérification des livraisons | |
|
232
|
7.4.1
|
S'en tenir à des réactions avec le fournisseur proportionnés au risque associé avec le produit acheté en cas de non-respect des spécifications d'achat | Et la satisfaction aux exigences réglementaires applicables. Cf. § 7.1 | |
|
233
|
7.4.1
|
Conserver les enregistrements des résultats de l'évaluation, sélection, surveillance et réévaluation de la capacité ou performance du fournisseur | Et des actions nécessaires entreprises. Cf. § 4.2.5 | |
|
|
7.4.2
|
Informations relatives aux achats
|
|
|
|
234
|
7.4.2
|
Décrire ou référencer le produit à acheter | Sont prises en compte toutes les données du produit acheté (spécifications, conditions de transport, d'emballage, de réception, de test, de stockage et autres) | |
| 235 | 7.4.2 a | Déterminer les spécifications du produit | Cf. § 7.2.1 | |
| 236 | 7.4.2 b | Déterminer les exigences d'acceptation, les procédures, processus et équipements | Par rapport au produit à acheter | |
| 237 | 7.4.2 c | Déterminer les exigences pour la qualification du personnel du fournisseur | Pour des cas spécifiques le personnel reçoit une formation appropriée (produit de type nouveau, machine ou équipement non utilisé jusque-là) | |
| 238 | 7.4.2 d | Déterminer les exigences relatives au SMQ | Tout ce qui concerne la maîtrise des non conformités liés au produit acheté et les actions, responsables et délais à mettre en place | |
| 239 |
7.4.2
|
S'assurer de l'adéquation des exigences d'achat spécifiées | Avant leur communication au fournisseur | |
| 240 |
7.4.2
|
Si applicable, établir avec le fournisseur un accord écrit lui imposant de notifier par avance les modifications apportées au produit acheté | Modifications qui peuvent impacter sur la satisfaction aux exigences d'achat spécifiées | |
|
241
|
7.4.2
|
Conserver des informations pertinentes relatives aux achats sous forme de documents et d'enregistrements dans la mesure requise pour la traçabilité | Cf. §§ 4.2.4, 4.2.5 et 7.5.9 | |
|
|
7.4.3
|
Vérification du produit acheté
|
||
|
242
|
7.4.3
|
Établir et mettre en place des activités d'inspection | Afin de s'assurer que le produit acheté satisfait aux exigences d'achat spécifiées. Le processus d'achat inclut l'identification et la mise en place des inspections en entrée et en cours de fabrication du produit | |
|
243
|
7.4.3
|
Fonder l'étendue des activités de vérification sur les résultats d'évaluation du fournisseur et proportionnée aux risques associés au produit acheté | Cf. § 7.1. Lien entre les activités de vérification et les résultats de l'évaluation du fournisseur et aussi le niveau de risque associé au produit acheté | |
|
244
|
7.4.3
|
Déterminer leur incidence sur le processus de réalisation du produit ou le dispositif médical en cas de modifications apportées au produit acheté | Cf. § 7.3.9 | |
|
245
|
7.4.3
|
Faire état des activités de vérification prévues et des modalités de libération du produit dans les informations relatives aux achats lorsque l'entreprise ou son client a l'intention d'effectuer des vérifications chez le fournisseur, | Sans acceptation écrite en amont de la part du fournisseur toute demande peut être refusée | |
|
246
|
7.4.3
|
Conserver les enregistrements de vérification | Cf. § 4.2.5 | |
|
|
Production et prestation de service
|
|||
|
|
Maîtrise de la production et de la prestation de service
|
|
||
| 247 | 7.5.1 | Planifier, réaliser, surveiller et maîtriser la production et la prestation de service | Afin d'assurer des produits conformes aux spécifications | |
| 248 | 7.5.1 a | Inclure une documentation de procédures et méthodes pour la maîtrise de la production | Cf. § 4.2.4 | |
| 249 |
7.5.1 b
|
Inclure la qualification des infrastructures | Cf. § 6.3 | |
| 250 | 7.5.1 c | Inclure la mise en place de la surveillance et le mesurage des paramètres des processus et des caractéristiques des produits | La maintenance des équipements est régulière | |
| 251 |
7.5.1 d
|
Inclure la disponibilité et l'utilisation des équipements de mesure | Les équipements de surveillance et de mesure sont maintenus en bon état et le personnel est formé à leur utilisation | |
| 252 | 7.5.1 e | Inclure la mise en place des opérations d'étiquetage et d'emballage | Conformément aux exigences relatives au produit, cf. § 7.2.1. Plus d'informations dans l'ISO 15223 (Dispositifs médicaux - Symboles à utiliser avec les étiquettes, l'étiquetage et les informations à fournir relatifs aux dispositifs médicaux -- Partie 1: Exigences générales) | |
| 253 |
7.5.1 f
|
Inclure la mise en place des activités de libération du produit, de livraison et de service après livraison | Les équipements de surveillance et de mesure sont utilisés régulièrement | |
| 254 |
7.5.1
|
Établir et tenir à jour un enregistrement pour tout dispositif médical ou lot de dispositifs médicaux | Cf. § 4.2.5. Pour fournir la traçabilité spécifiée. Cf. § 7.5.9 | |
| 255 | 7.5.1 | Établir et tenir à jour un enregistrement pour identifier la quantité fabriquée et la quantité approuvée pour distribution | Cf. § 4.2.5 | |
| 256 | 7.5.1 | Vérifier et approuver chaque enregistrement | Cf.§ 4.2.5 | |
|
Propreté du produit
|
|
|||
| 257 | 7.5.2 a | Documenter les exigences relatives à la propreté ou la maîtrise de la contamination quand le produit est nettoyé par l'entreprise avant sa stérilisation ou son utilisation | Cf. § 4.2.4 | |
|
258
|
7.5.2 b
|
Documenter les exigences relatives à la propreté ou la maîtrise de la contamination quand le produit est fourni à l'état non stérile | Et le produit est soumis à un processus de nettoyage avant sa stérilisation ou son utilisation. Cf. § 4.2.5 | |
|
259
|
7.5.2 c
|
Documenter les exigences relatives à la propreté ou la maîtrise de la contamination quand le produit ne peut être nettoyé avant sa stérilisation ou son utilisation | Et la propreté du produit est importante en utilisation. Cf. § 4.2.4 | |
| 260 | 7.5.2 d | Documenter les exigences relatives à la propreté ou la maîtrise de la contamination quand le produit est fourni pour être utilisé à l'état non stérile | Et la propreté du produit est importante en utilisation. Cf. § 4.2.4 | |
| 261 |
7.5.2 e
|
Documenter les exigences relatives à la propreté ou la maîtrise de la contamination quand des agents de traitement doivent être éliminés du produit pendant sa fabrication | Cf. § 4.2.4 | |
| 262 | 7.5.2 | Ne pas appliquer les exigences avant le processus de nettoyage si le produit est nettoyé conformément à a) ou b) ci-dessus | Cf. § 6.4.1 | |
|
|
Activités d'installation
|
|||
| 263 | 7.5.3 | Documenter les exigences des critères d'acceptation et de vérification d'installation du dispositif médical, si approprié | Cf. § 4.2.4 | |
| 264 | 7.5.3 | Fournir les exigences documentées pour l'installation et la vérification d'installation du dispositif médical | Si les exigences du client convenues permettent une installation par un prestataire externe en dehors en dehors de l'entreprise ou son fournisseur | |
| 265 | 7.5.3 | Conserver les enregistrements de l'installation et de la vérification du dispositif médical réalisées par l'entreprise ou son fournisseur | Cf. § 4.2.5 | |
|
|
Prestations associées
|
|||
|
266
|
7.5.4
|
Documenter des procédures, des instructions de travail et des références de mesurage pour la réalisation des prestations associées et la vérification que les exigences du produit sont satisfaites | f. § 4.2.4. Si les prestations associées du dispositif médical constituent une exigence spécifiée (comme par exemple la maintenance) | |
| 267 | 7.5.4 a | Analyser les enregistrements des prestations associées | Afin de déterminer si ces informations doivent être considérées comme une réclamation | |
| 268 | 7.5.4 b | Analyser les enregistrements des prestations associées | Afin d'utiliser ces informations comme éléments d'entrée du processus d'amélioration | |
| 269 | 7.5.4 | Conserver les enregistrements des prestations associées réalisées | Cf. § 4.2.5 | |
|
|
Exigences particulières pour les dispositifs médicaux stériles
|
|
||
| 270 | 7.5.5 | Conserver les enregistrements des paramètres de stérilisation pour chaque lot de stérilisation | Cf. § 4.2.5 | |
| 271 | 7.5.5 | Permettre de garantir la traçabilité de chaque lot de production des dispositifs médicaux | Avec les enregistrements de stérilisation. Cf. § 7.5.9 | |
|
|
Validation des processus de production et de prestation de service
|
|
||
| 272 | 7.5.6 | Valider tout processus de production et de prestation de service dont les éléments de sortie ne peuvent être vérifiés par une surveillance ou mesure effectuées a posteriori | Ceci inclut en conséquence tous les processus pour lesquels des déficiences n'apparaissent qu'une fois le produit en usage ou le service livré | |
| 273 | 7.5.6 | Démontrer par la validation l'aptitude de ces processus à toujours atteindre les résultats planifiés | La validation est réalisée après la revue et la vérification des processus | |
| 274 | 7.5.6 a | Documenter des procédures pour la validation des processus incluant des critères définis pour leur revue et approbation | Cf. § 4.2.4 | |
| 275 | 7.5.6 b | Documenter des procédures pour la validation des processus incluant l'approbation des équipements | Et la qualification du personnel. Cf. § 4.2.4 | |
| 276 | 7.5.6 c | Documenter des procédures pour la validation des processus incluant l'utilisation de méthodes spécifiques, de procédures et critères d'acceptation | Cf. § 4.2.4 | |
| 277 | 7.5.6 d | Documenter des procédures pour la validation des processus incluant des techniques statistiques avec justification de la taille d'échantillonnage | Selon le cas, cf. § 4.2.4 | |
| 278 | 7.5.6 e | Documenter des procédures pour la validation des processus incluant des exigences pour les enregistrements | Cf. §§ 4.2.4 et 4.2.5 | |
| 279 | 7.5.6 f | Documenter des procédures pour la validation des processus incluant la revalidation | Cf. § 4.2.4. Y compris les critères de revalidation | |
| 280 | 7.5.6 g | Documenter des procédures pour la validation des processus incluant l'approbation des modifications apportées au processus | Cf. § 4.2.4 | |
| 281 | 7.5.6 | Documenter des procédures de validation des applications logicielles utilisées en production et en prestation de service | Cf. § 4.2.4 | |
| 282 | 7.5.6 | Valider les applications logicielles avant leur première utilisation | Et aussi après modification du logiciel ou de son application | |
| 283 | 7.5.6 | Mettre en place une démarche proportionnée au risque associé à son utilisation, y compris son incidence sur l’aptitude du produit à respecter les spécifications | Cf. § 7.1 | |
| 284 | 7.5.6 | Conserver les enregistrements des résultats et conclusions de validation et des actions nécessaires entreprises | Cf. §§ 4.2.4 et 4.2.5 | |
|
|
Exigences spécifiques relatives à la validation des procédés de stérilisation et des systèmes de barrière stérile
|
|
||
| 285 | 7.5.7 | Documenter des procédures pour la validation des procédés de stérilisation et des systèmes de barrière stérile | Cf. § 4.2.4 | |
| 286 | 7.5.7 | Valider les procédés de stérilisation et les systèmes de barrière stérile avant leur mise en œuvre | Et suite aux modifications apportées au produit ou au procédé, selon le cas | |
| 287 | 7.5.7 | Conserver les enregistrements des résultats et conclusions de validation et des actions nécessaires entreprises | Cf. §§ 4.2.4 et 4.2.5. Plus d'informations dans l'ISO 11607-1 (Emballages des dispositifs médicaux stérilisés au stade terminal - Partie 1: Exigences relatives aux matériaux, aux systèmes de barrière stérile et aux systèmes d'emballage) et l'ISO 11607-2 (Emballages des dispositifs médicaux stérilisés au stade terminal -- Partie 2: Exigences de validation pour les procédés de formage, scellage et assemblage) | |
|
|
7.5.8
|
Identification
|
|
|
| 288 | 7.5.8 | Documenter des procédures d'identification du produit | Cf. § 4.2.4 | |
| 289 | 7.5.8 | Identifier le produit à l'aide de moyens adaptés | Tout au long de sa réalisation | |
| 290 | 7.5.8 | Identifier l'état du produit conformément aux exigences de surveillance et mesurage | Tout au long de la réalisation du produit | |
| 291 | 7.5.8 | Tenir à jour l'identification de l'état du produit tout au long de la réalisation du produit, du stockage, de l'installation et des prestations associées | Afin de s'assurer que seul un produit ayant passé avec succès les inspections et tests ou libéré avec une dérogation autorisée est diffusé, utilisé ou installé | |
| 292 | 7.5.8 | Documenter un système pour attribuer un identifiant unique au dispositif médical | Cf. § 4.2.5. Si requis par les exigences réglementaires applicables | |
| 293 | 7.5.8 | Documenter des procédures pour s'assurer que les dispositifs médicaux retournés sont identifiés et distingués du produit conforme | Cf. § 4.2.4 | |
|
|
Traçabilité
|
|
||
|
|
7.5.9.1
|
Généralités
|
|
|
| 294 | 7.5.9.1 | Documenter des procédures pour la traçabilité | Cf. § 4.2.4. La préservation du produit comprend toutes les étapes du cycle de vie du produit (réception, production, manutention, stockage, livraison) | |
| 295 | 7.5.9.1 | Définir l'étendue de la traçabilité du produit | Conformément aux exigences réglementaires applicables | |
| 296 | 7.5.9.1 | Conserver les enregistrements de traçabilité | Cf. § 4.2.5 | |
|
|
7.5.9.2
|
Exigences particulières pour des dispositifs médicaux implantables
|
|
|
| 297 | 7.5.9.2 | Inclure dans les enregistrements nécessaires à la traçabilité tous les composants, matériaux et conditions d'environnement de travail utilisés | Lorsque ces paramètres peuvent entraîner une non-conformité du dispositif médical aux exigences de sécurité et de performance spécifiées | |
| 298 | 7.5.9.2 | Exiger que ses agents ou distributeurs conservent des enregistrements de la distribution de dispositifs médicaux en vue d'en assurer la traçabilité | Et que ces enregistrements soient accessibles pour inspection | |
| 299 | 7.5.9.2 | S'assurer que les coordonnées du destinataire du colis d'expédition sont enregistrées | Nom et adresse. Cf. § 4.2.5 | |
|
|
7.5.10
|
Propriété du client
|
|
|
| 300 | 7.5.10 | Identifier, vérifier, protéger et sauvegarder la propriété que le client a fournie pour être utilisée ou incorporée dans le produit | Lorsque la propriété du client se trouve sous le contrôle de l'entreprise ou qu'elle l'utilise | |
| 301 | 7.5.10 | Notifier le client | Si la propriété du client est perdue, endommagée ou encore jugée impropre à l'utilisation | |
| 302 | 7.5.10 | Conserver les enregistrements des notifications | Cf. § 4.2.5 | |
|
|
7.5.11
|
Préservation du produit
|
|
|
| 303 | 7.5.11 | Documenter des procédures pour la préservation de la conformité du produit pendant les opérations internes, le stockage, la manipulation et la distribution | Cf. § 4.2.4. La préservation du produit comprend toutes les étapes du cycle de vie du produit (réception, production, manutention, stockage, livraison, distribution) | |
|
304
|
7.5.11
|
Appliquer la préservation aux composants du dispositif médical | Y compris l'emballage | |
| 305 | 7.5.11 | Protéger le produit contre tous dommages, altérations ou contaminations quand exposé aux conditions et dangers attendus pendant le traitement, le stockage, la manutention et la distribution | Au moyen de la conception et la construction d'emballages et conteneurs d’expédition adaptés | |
| 306 | 7.5.11 | Protéger le produit contre tous dommages, altérations ou contaminations quand exposé aux conditions et dangers attendus pendant le traitement, le stockage, la manutention et la distribution | Au moyen de la documentation des exigences pour les conditions particulières nécessaires, si la préservation ne peut être assurée par l’emballage | |
| 307 | 7.5.11 | Maîtriser les conditions spéciales requises | Comme durée de conservation limitée et conditions de stockage particulières | |
| 308 | 7.5.11 | Enregistrer les conditions spéciales requises | Cf. § 4.2.5 | |
|
|
Maîtrise des équipements de surveillance et de mesure
|
|
||
|
309
|
7.6
|
Déterminer les activités de surveillance et de mesure à entreprendre | Les processus de surveillance et de mesure sont en place pour apporter des preuves de la conformité du produit. Cf. § 7.2.1 | |
| 310 | 7.6 | Déterminer les équipements de surveillance et de mesure | Maintenir une liste des équipements | |
| 311 | 7.6 | Documenter des procédures pour assurer que les activités de surveillance et de mesure peuvent être effectuées et sont effectuées de manière cohérente | Cf. § 4.2.4. Par rapport aux exigences de surveillance et de mesure | |
| 312 | 7.6 a | Pour des résultats valables s'assurer que les équipements de mesure sont étalonnés ou vérifiés à intervalles spécifiés ou avant leur utilisation | Par rapport à des étalons de mesure reliés à des étalons de mesure internationaux ou nationaux. Lorsque ces étalons n'existent pas, la référence utilisée pour l'étalonnage ou la vérification est enregistrée. Cf. § 4.2.5 | |
|
313
|
7.6 b
|
S'assurer que les équipements de mesure sont réglés ou réglés de nouveau | Les équipements de mesure sont réglés régulièrement | |
|
314
|
7.6 b
|
Enregistrer les réglages effectués | Cf. § 4.2.5 | |
| 315 |
7.6 c
|
S'assurer que les équipements de mesure sont identifiés | Pour pouvoir déterminer l'état d'étalonnage | |
|
316
|
7.6 d
|
S'assurer que les équipements de mesure sont protégés contre des réglages susceptibles d'invalider le résultat de la mesure | Protection efficace non seulement pendant leur utilisation (déplacement, maintenance, stockage) | |
|
317
|
7.6 e
|
S'assurer que les équipements de mesure sont protégés contre tous dommages et détériorations au cours de leur manutention, maintenance et stockage | Cela concerne des produits potentiellement non-conformes | |
| 318 |
7.6
|
Procéder à l’étalonnage ou la vérification selon la procédure documentée | L'équipement est vérifié et étalonné. Les produits sont inspectés, validés (avec ou sans dérogation) ou identifiés comme non-conformes | |
|
319
|
7.6
|
Évaluer et enregistrer la validité des résultats de mesure antérieurs | Lorsque l'équipement se révèle non conforme aux exigences | |
|
320
|
7.6
|
Entreprendre les actions appropriées sur l'équipement non conforme et tout produit affecté | Les logiciels de surveillance et de mesure sont validés préalablement à leur utilisation | |
|
321
|
7.6
|
Conserver les enregistrements des résultats d'étalonnage et de vérification | Cf. § 4.2.5 | |
|
322
|
7.6
|
Documenter des procédures pour la validation de l'application des logiciels utilisés pour la surveillance et la mesure des exigences | Cf. § 4.2.4. Effectuer une deuxième validation si la première n'est pas satisfaisante | |
|
323
|
7.6
|
Valider les logiciels avant leur première utilisation et après modification du logiciel ou de son application, selon le cas | Effectuer une deuxième validation si la première n'est pas satisfaisante | |
|
324
|
7.6
|
Définir l'approche et les activités pour la validation et la revalidation des logiciels proportionnées au risque associé à l’utilisation du logiciel | Y compris l'effet de la capacité du produit être conforme aux spécifications. Cf. § 7.1 | |
|
325
|
7.6
|
Conserver les enregistrements des résultats et des conclusions de validation et des actions nécessaires entreprises | Cf. §§ 4.2.4 et 4.2.5. Plus d'informations dans l'ISO 10012 (Systèmes de management de la mesure - Exigences pour les processus et les équipements de mesure) | |
|
|
Mesurage, analyse et amélioration
|
|||
|
|
Généralités
|
|
||
|
326
|
8.1 a
|
Planifier et mettre en œuvre les processus de surveillance, mesurage, analyse et amélioration nécessaires | Afin de démontrer la conformité du produit. La maîtrise de la conformité du produit est démontrée par des processus d'inspection tout au long des étapes de production | |
| 327 | 8.1 b | Assurer la conformité du système de management de la qualité | "Si vous ne pouvez le mesurer, vous ne pouvez le maîtriser. Peter Drucker" | |
| 328 |
8.1 c
|
Maintenir l'efficacité du SMQ | La maîtrise de la conformité du SMQ est assurée entre autres par des processus de management (stratégie, audit, amélioration continue, autoévaluation) | |
| 329 | 8.1 | Inclure la détermination des méthodes applicables | Y compris les techniques statistiques et l'étendue de leur utilisation | |
|
|
Surveillance et mesurage
|
|
||
|
|
8.2.1 |
Retours d'information
|
|
|
|
330
|
8.2.1
|
Recueillir et surveiller les informations relatives au niveau de satisfaction aux exigences du client | Comme une des mesures de la performance du SMQ | |
| 331 | 8.2.1 | Documenter la méthode d'obtention et d'usage de ces informations | Cf. § 4.2.5 | |
| 332 | 8.2.1 | Documenter des procédures pour les processus de retour d'information | Cf. §§ 4.2.4 et 7.2.3 | |
|
333
|
8.2.1
|
Inclure dans ce processus de retour d'information les dispositions pour recueillir des données de la production | Et des activités post-production | |
| 334 | 8.2.1 | Utiliser les informations recueillies par le processus de retour d'information comme donnée d'entrée potentielle pour la gestion des risques | Afin de surveiller et maintenir les exigences du produit, la réalisation du produit et les processus d'amélioration. Cf. § 7.1 | |
|
335
|
8.2.1
|
Inclure dans le processus de retour d'information la revue des enseignements des activités post-production | Si cela est demandé par les exigences réglementaires applicables | |
|
|
8.2.2 |
Traitement des réclamations
|
|
|
|
336
|
8.2.2
|
Documenter des procédures pour le traitement des réclamations | Cf. § 4.2.4. Dans des délais appropriés conformément aux exigences réglementaires applicables | |
|
337
|
8.2.2 a
|
Inclure des exigences et des responsabilités pour réceptionner et enregistrer les informations | Qui, quand, comment | |
|
338
|
8.2.2 b
|
Évaluer l'information pour déterminer si le retour d'information est une réclamation | Par la personne en charge et son équipe | |
|
339
|
8.2.2 c
|
Enquêter les réclamations | La méthode 8 D est judicieuse pour répondre à cette exigence | |
|
340
|
8.2.2 d
|
Déterminer le besoin de signaler aux autorités réglementaires appropriées | Cf. § 8.2.3 | |
|
341
|
8.2.2 e
|
Traiter le produit en rapport avec la réclamation | Vérifier l'étendue de la réclamation | |
|
342
|
8.2.2 f
|
Déterminer le besoin d'entreprendre des corrections et des actions correctives | Et entreprendre les actions nécessaires | |
|
343
|
8.2.2
|
Documenter une justification si la réclamation n'est pas enquêtée | Cf. § 4.2.4 | |
|
344
|
8.2.2
|
Documenter toute correction ou action corrective suite à une réclamation | Cf. § 4.2.4 | |
|
345
|
8.2.2
|
Échanger des informations pertinentes avec la partie externe | Lorsque l'enquête détermine que des activités en dehors de l'entreprise ont contribué à la réclamation | |
|
346
|
8.2.2
|
Conserver les enregistrements du traitement des réclamations | Cf. § 4.2.5 | |
| 8.2.3 |
Signalement aux autorités réglementaires
|
|
||
| 347 |
8.2.3
|
Documenter des procédures pour le signalement aux autorités réglementaires appropriées des événements répondant aux critères de signalement spécifiés ou la diffusion d’une fiche d’avertissement | Cf. § 4.2.4. Si cela est requis par les exigences réglementaires applicables | |
|
348
|
8.2.3
|
Conserver des enregistrements du signalement aux autorités réglementaires | Cf. § 4.2.5 | |
|
|
8.2.4 |
Audit interne
|
|
|
| 349 |
8.2.4 a
|
Mener des audits internes à intervalles planifiés | Afin de déterminer si le SMQ est conforme aux dispositions planifiées et documentées, aux exigences de la norme ISO 13485, aux exigences du SMQ établies par l'entreprise et aux exigences réglementaires applicables | |
|
350
|
8.2.4 b
|
Déterminer si le SMQ est mis en œuvre et entretenu de manière efficace | Cf. § 7.1 | |
|
351
|
8.2.4
|
Documenter des procédures pour décrire les responsabilités et exigences | Cf. § 4.2.4. Afin de planifier et mener des audits, enregistrer et rendre compte des résultats des audits | |
|
352
|
8.2.4
|
Planifier un programme d'audit en tenant compte de l'état et de l'importance des processus et des domaines à auditer | Y compris les résultats des audits précédents | |
|
353
|
8.2.4
|
Définir et enregistrer les critères, le champ, la fréquence et les méthodes d'audit | Cf. § 4.2.5 | |
|
354
|
8.2.4
|
Assurer l'objectivité et l'impartialité du processus d'audit | Avec le choix des auditeurs et la réalisation des audits | |
|
355
|
8.2.4
|
Assurer que les auditeurs n'auditent pas leur propre travail | ||
|
356
|
8.2.4
|
Conserver les enregistrements des audits et leurs résultats | Cf. § 4.2.5. Y compris l'identification des processus, des champs audités et les conclusions | |
|
357
|
8.2.4
|
Assurer par l'encadrement responsable du domaine audité que des actions sont entreprises sans délai indu | Afin d'éliminer les non-conformités détectées et leurs causes | |
|
358
|
8.2.4
|
Inclure dans les activités de suivi la vérification des actions entreprises et le compte rendu des résultats de cette vérification | Plus d'information dans l'ISO 19011 (Lignes directrices pour l'audit des systèmes de management) | |
| 8.2.5 |
Surveillance et mesure des processus
|
|
||
| 359 |
8.2.5
|
Utiliser des méthodes appropriées pour la surveillance | Et, lorsqu'elle est applicable, la mesure des processus du SMQ | |
|
360
|
8.2.5
|
Démontrer avec ces méthodes l'aptitude des processus à atteindre les résultats planifiés | Cf. § 7.1 | |
|
361
|
8.2.5
|
Lorsque les résultats planifiés ne sont pas atteints entreprendre des corrections et des actions correctives, comme il convient | Cf. § 8.5.2 | |
|
|
8.2.6 |
Surveillance et mesure du produit
|
|
|
| 362 |
8.2.6
|
Surveiller et mesurer les caractéristiques du produit | Afin de vérifier que les exigences relatives au produit ont été satisfaites | |
|
363
|
8.2.6
|
Effectuer ceci à des étapes appropriées du processus de réalisation du produit | Conformément aux dispositions planifiées et aux procédures documentées | |
|
364
|
8.2.6
|
Conserver la preuve de la conformité aux critères d'acceptation | Cf. § 4.2.5 | |
|
365
|
8.2.6
|
Enregistrer l'identité de la personne ayant autorisé la mise à disposition du produit | Cf. § 4.2.5 | |
|
366
|
8.2.6
|
Identifier dans les enregistrements les équipements d’essai utilisés pour effectuer les mesures | Si approprié, cf. § 4.2.5 | |
|
367
|
8.2.6
|
Effectuer la libération du produit et la prestation du service avant l'exécution satisfaisante des dispositions planifiées et documentées | Cf. § 7.1 | |
|
368
|
8.2.6
|
Enregistrer l'identité des personnes chargées d'effectuer une inspection ou un essai pour des dispositifs médicaux implantables | Cf. § 4.2.5 | |
| 8.3 |
Maîtrise du produit non conforme
|
|
||
|
|
8.3.1 |
Généralités
|
|
|
| 369 |
8.3.1
|
Assurer que le produit non conforme est identifié et maîtrisé | Afin d'empêcher son utilisation ou sa livraison | |
|
370
|
8.3.1
|
Documenter une procédure pour définir la maîtrise du produit non conforme | Cf. § 4.2.4. Déterminer les responsabilités et autorités associées, l'identification, la documentation, l'isolement, l'évaluation et le traitement des non-conformités | |
|
371
|
8.3.1
|
Inclure dans l'évaluation de la non-conformité la détermination du besoin d'enquête | Et le signalement à toute partie externe responsable de la non-conformité | |
|
372
|
8.3.1
|
Conserver les enregistrements de la nature des non-conformités et des actions entreprises | Cf. § 4.2.5. Y compris l'évaluation, toute enquête déclenchée et la justification des décisions | |
| 8.3.2 |
Actions en réponse à une non-conformité du produit détectée avant livraison
|
|
||
| 373 |
8.3.2 a
|
Mener des actions permettant d'éliminer la non-conformité détectée | Pour traiter le produit non conforme | |
|
374
|
8.3.2 b
|
Mener des actions permettant d'empêcher son utilisation ou application prévue | Pour traiter le produit non conforme | |
|
375
|
8.3.2 c
|
Autoriser son utilisation, sa libération ou son approbation par dérogation | Pour traiter le produit non conforme | |
|
376
|
8.3.2
|
S'assurer que le produit non conforme n'est accepté par dérogation que si la justification est fournie | Et aussi que l'approbation est obtenue et les exigences réglementaires applicables sont satisfaites | |
|
377
|
8.3.2
|
Conserver l'acceptation de la dérogation et l'identité de la personne autorisant la dérogation | Cf. § 4.2.5 | |
|
|
8.3.3 |
Actions en réponse à une non-conformité du produit détectée après livraison
|
|
|
| 378 |
8.3.3
|
Mener des actions adaptées aux effets, réels ou potentiels, de la non-conformité | Afin de traiter le produit non conforme détecté après livraison ou après que son utilisation a commencé | |
|
379
|
8.3.3
|
Conserver les enregistrements des actions entreprises | Cf. § 4.2.5 | |
|
380
|
8.3.3
|
Documenter la procédure pour diffuser des fiches d'avertissement conformément aux exigences réglementaires applicables | Cf. § 4.2.4 | |
|
381
|
8.3.3
|
Etre capable à tout moment de déclencher ces procédures | Afin de diffuser les fiches d'avertissement sans délai | |
|
382
|
8.3.3
|
Conserver les enregistrements des actions liées à la diffusion des fiches d'avertissement | Cf. § 4.2.5 | |
| 8.3.4 |
Retouches
|
|
||
| 383 |
8.3.4
|
Effectuer des retouches conformément aux procédures documentées | Cf. § 4.2.4. Prendre en compte l'effet négatif potentiel de la retouche sur le produit | |
|
384
|
8.3.4
|
Soumettre ces procédures à la même revue et approbation que la procédure originale | Remettre dans le flux normal | |
|
385
|
8.3.4
|
Vérifier le produit retouché | Pour s'assurer qu'il satisfait aux critères d'acceptation et les exigences réglementaires | |
|
386
|
8.3.4
|
Conserver les enregistrements des retouches | Cf. § 4.2.5 | |
|
|
Analyse des données
|
|
||
| 387 |
8.4
|
Documenter la procédure pour déterminer, recueillir et analyser les données appropriées | Cf. § 4.2.4. Afin de démontrer la pertinence, l'adéquation et l'efficacité du SMQ | |
| 388 |
8.4
|
Inclure dans la procédure la détermination des méthodes appropriées | Y compris les techniques statistiques et l'étendue de leur utilisation | |
|
389
|
8.4
|
Inclure dans l'analyse des données les données résultant des activités de surveillance et de mesure | Et d'autres sources pertinentes | |
|
390
|
8.4 a
|
Inclure les informations sur les retours d'information | Cf. § 8.2.1 | |
|
391
|
8.4 b
|
Inclure les informations sur la satisfaction aux exigences du produit | Cf. § 7.2.1 | |
|
392
|
8.4 c
|
Inclure les informations sur les caractéristiques et tendances des processus et produits | Y compris les opportunités d'amélioration | |
|
393
|
8.4 d
|
Inclure les informations sur les fournisseurs | Cf. § 7.4.1 | |
|
394
|
8.4 e
|
Inclure les informations sur les audits | Cf. § 8.2.4 | |
|
395
|
8.4 f
|
Inclure les informations sur les comptes rendus des prestations de service | Selon le cas, cf. § 7.5.4 | |
|
396
|
8.4
|
Utiliser cette analyse comme élément d’entrée d’amélioration | Si l’analyse de données montre que le SMQ n'est pas approprié, adéquat ou efficace. Cf. § 8.5 | |
|
397
|
8.4
|
Conserver les enregistrements des résultats des analyses | Cf. § 4.2.5 | |
|
|
Amélioration
|
|
||
|
|
8.5.1 |
Généralités
|
|
|
| 398 |
8.5.1
|
Identifier et mettre en œuvre toutes les modifications nécessaires pour assurer et maintenir la pertinence, l'adéquation et l'efficacité permanentes du SMQ et la sécurité et les performances du dispositif médical | En utilisant la politique qualité, les objectifs qualité, les résultats d'audits, la surveillance après mise sur le marché, l'analyse des données, les actions correctives et préventives ainsi que la revue de direction | |
|
|
8.5.2 |
Actions correctives
|
|
|
| 399 |
8.5.2
|
Mener des actions pour éliminer les causes de non-conformités | Afin d'éviter toute réapparition | |
| 400 |
8.5.2
|
Entreprendre sans délai toutes les actions correctives nécessaires | Ne pas attendre est fondamental | |
| 401 |
8.5.2
|
Adapter les actions correctives aux effets des non-conformités | La procédure pour les actions correctives répond aux questions qui, quand, comment, dans quelles conditions, avec quelles ressources identifier et traiter les non-conformités, déterminer et éliminer les causes, évaluer le besoin d'actions correctives, mettre en place les actions correctives et passer en revue les actions | |
| 402 |
8.5.2 a
|
Documenter la procédure actions correctives pour définir les exigences pour passer en revue les non-conformités | Cf. § 4.2.4. Inclure dans la procédure la méthode pour trouver les causes des non-conformités (diagramme d'Ishikawa, maîtrise statistique des processus) | |
| 403 |
8.5.2 b
|
Déterminer les causes de non-conformités | Utiliser la méthode des 5 P (pourquoi) | |
|
404
|
8.5.2 c
|
Évaluer le besoin d'entreprendre des actions | Afin de s'assurer que les non-conformités ne se reproduisent pas | |
|
405
|
8.5.2 d
|
Planifier, documenter et mettre en œuvre les actions nécessaires | Cf. § 4.2.4. Y compris, selon le cas, mettre à jour la documentation | |
|
406
|
8.5.2 e
|
Vérifier que l’action corrective n’a pas d’effet négatif | Sur la capacité à satisfaire aux exigences réglementaires applicables ou sur la sécurité et les performances du dispositif médical | |
|
407
|
8.5.2 f
|
Passer en revue l'efficacité des actions correctives entreprises | Afin de s'assurer que toute réapparition est exclue | |
|
408
|
8.5.2
|
Conserver les enregistrements de toute enquête et action entreprise | Cf. § 4.2.5 | |
|
|
Actions préventives
|
|
||
| 409 |
8.5.3
|
Déterminer les actions permettant d’éliminer les causes de non-conformités potentielles | Afin d'éviter toute apparition. Analyse et éradication des causes potentielles des non-conformités des processus ou du SMQ | |
| 410 |
8.5.3
|
Adapter les actions préventives aux effets des problèmes potentiels | Faire le nécessaire par rapport aux effets potentiels (rester dans la limite des ressources disponibles) | |
| 411 |
8.5.3 a
|
Documenter la procédure actions préventives pour décrire les exigences pour déterminer les non-conformités potentielles et leurs causes | Cf. § 4.2.4. La procédure pour les actions préventives répond aux questions qui, quand, comment, dans quelles conditions, avec quelles ressources identifier les non-conformités potentielles, déterminer leurs causes, évaluer le besoin d'actions préventives, mettre en place les actions préventives et passer en revue les actions | |
| 412 |
8.5.3 b
|
Évaluer le besoin d'entreprendre des actions | Afin de s'assurer que les non-conformités ne se produisent pas. L'apparition sera-t-elle évitée ? Les ressources nécessaires sont-elles à disposition ? | |
| 413 |
8.5.3 c
|
Planifier, documenter et mettre en œuvre les actions nécessaires | Cf. § 4.2.4. Y compris, selon le cas, mettre à jour la documentation | |
| 414 | 8.5.3 d | Vérifier que l’action préventive n’a pas d’effet négatif | Sur la capacité à satisfaire aux exigences réglementaires applicables ou sur la sécurité et les performances du dispositif médical | |
|
415
|
8.5.3 e
|
Passer en revue l'efficacité des actions préventives entreprises | "La prévention coûte toujours moins cher" | |
|
416
|
8.5.3
|
Conserver les enregistrements de toute enquête et action entreprise | Cf. § 4.2.5 | |
|
|
|
|
|
|

